
Remember me
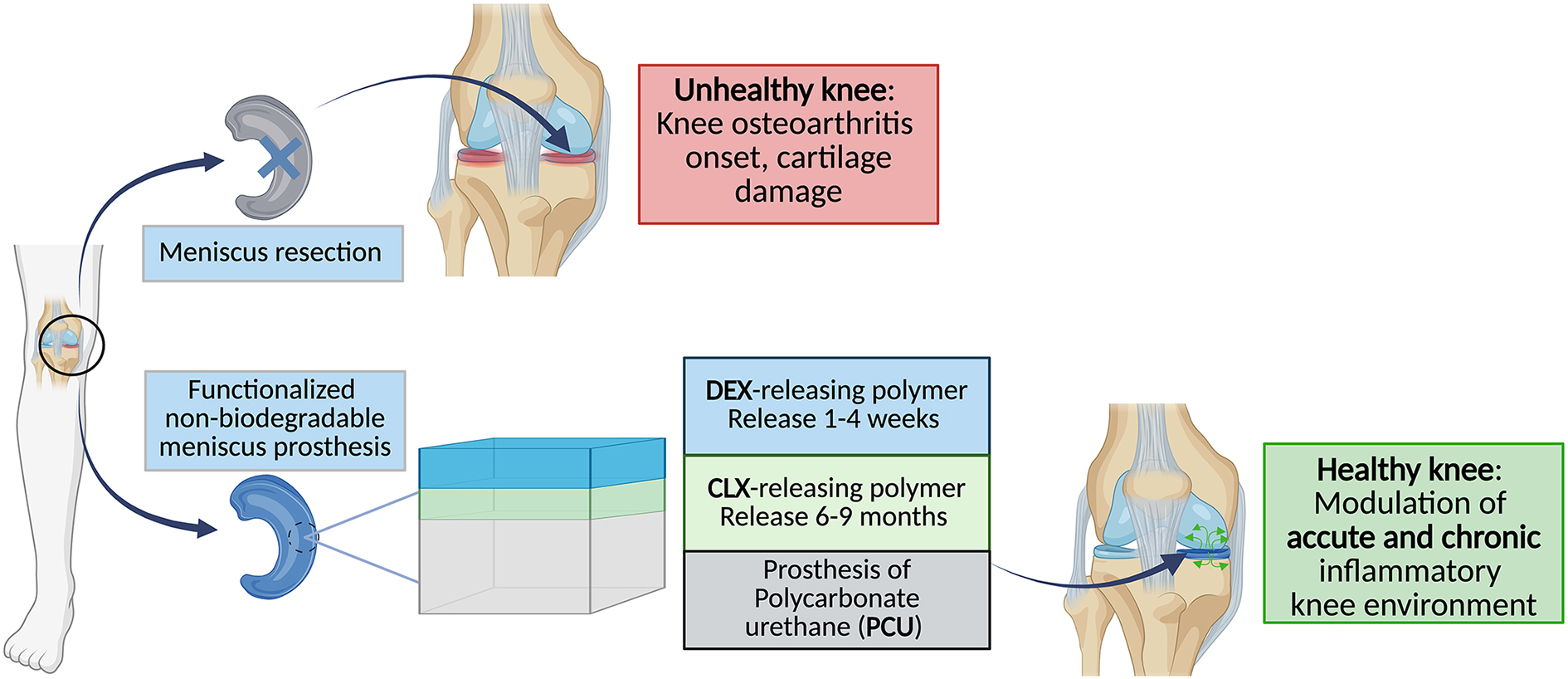
Our aim was to engineer a biodegradable bilayer polymer coating around a polycarbonate urethane (PCU) meniscus prosthesis to release two anti-inflammatory drugs with controlled release kinetics. More precisely, dexamethasone (DEX) was intended to be released within 1–4 weeks to alleviate the acute inflammation caused mainly by the surgery. Meanwhile, celecoxib (CLX) was intended to be released within 6–9 months to relieve the chronic inflammation related to the prosthesis. Both drugs are expected to mediate the incorporation of the implant into the knee cavity, reducing potential adverse inflammatory reactions. Therefore, we developed and optimized the preparation of the bilayer coating, and we performed experiments to evaluate the activity of released drugs in vitro. For this, we followed a specific work plan consisting of:
1)The screening of different biodegradable and biocompatible polymers for the independent release of DEX and CLX with the desired release kinetics.
2)The development of drug-releasing bilayer polymer coatings results from the combination of the most promising prototypes selected for the independent release of the drugs. This development consisted of evaluating the capacity of the bilayer systems to release CLX and DEX in a required time frame, characterization of their physicochemical properties, and in vitro study of their degradation.
3)The in vitro evaluation of the sterility and biocompatibility of the drugs and polymer degradation products from the bilayer coated system, and the anti-inflammatory activity of these drugs in human primary macrophages.
Design and optimization of drug-loaded polymer filmsPolyesters were selected as the base polymers of the bilayer coating due to their controlled biodegradability, tunable release properties and favorable regulatory profile. The primary objective of this section was to evaluate the influence of the polymer type on the drug release profile while keeping other parameters—such as the preparation method and the number of polymer layers—constant. Although the ultimate goal was to achieve the controlled release of two different anti-inflammatory drugs from the same meniscus prosthesis with distinct release kinetics, the initial screening focused on evaluating the release of each drug independently within a single polymeric matrix.
CLX-loaded polymer filmsPoly(caprolactone) (PCL) and poly(L-lactide) (PLLA) were chosen due to their slow degradation rate and, hence, long-term release capacity of CLX (6 to 9 months). This profile was expected to modulate a process of chronic inflammation [36, 37]. The influence of molecular weight (MW) on degradation rate was explored by testing high molecular weight (HMW) (~ 210 kDa HMW-PLLA and ~ 73 kDa HMW-PCL) and low molecular weight (LMW) (~ 74 kDa LMW-PLLA and ~ 32 kDa LMW-PCL) variants of each polymer [38,39,40,41,42]. CLX-loaded films were fabricated by dissolving CLX and each polymer in dichloromethane (DCM) and casting the solution onto square-shaped PCU implants. After curing, the films and PCU implants were immersed in PBS with 1% (w/v) Tween®80, and CLX release was monitored via UPLC (Fig. 1). The drug release study was halted when a plateau release phase was observed.
The release studies revealed that CLX was consistently released faster from PCL films, irrespective of MW, as compared to from PLLA films. PLLA exhibited MW-dependent release kinetics, with HMW-PLLA providing more sustained release compared to LMW-PLLA [40]. The differences in release kinetics can be explained by the polymer chain length. Since none of these polymers is expected to degrade within a few weeks, the observed release behavior aligns with previous studies linking polymer chain length to drug diffusion [43]. Moreover, this faster release may also be explained by increased polymer swelling and water uptake in LMW systems, which create transient pathways for drug diffusion due to reduced chain entanglement and matrix stiffness [44]. The denser matrix created by HMW-PLLA restricted water penetration, slowing degradation and drug diffusion and, therefore, delaying drug release. As expected, due to the larger difference in MW between variants, these effects are more pronounced in PLLA films (~ 140 kDa) than in PCL films (~ 41 kDa). In addition to polymer chain length, the degree of crystallinity also affects water uptake and drug diffusion. Higher crystallinity, as observed in HMW-PLLA and HMW-PCL, is associated with lower water permeability and slower drug release.
The plateau in release profiles is hypothesized to be a result of the interplay between drug diffusion and polymer degradation. Initially, the release is supposed to be diffusion-driven through water-filled pores, whereas the second phase corresponds to the release of the drug remaining trapped within the polymer matrix. Since polymer degradation is slow, this trapped drug remains unavailable, leading to incomplete release within the study timeframe. Thus, the release kinetics reflect both initial diffusion and delayed drug release associated with polymer degradation [45].
Although the release profile of HMW-PLLA might be considered aligned with the targeted release timeframe (6–9 months), the plateau observed after 20 weeks led to an incomplete drug release (30% of the total CLX released (Fig. 1)). This incomplete release was attributed to limited polymer degradation, which could lead to prototype limitations such as drug recrystallization [42]. Although recrystallization was not observed or specifically analyzed in this study, prolonged entrapment of drug molecules within the polymer matrix under aqueous conditions is generally considered a risk factor for potential recrystallization in drug delivery systems [46].
Fig. 1
Release kinetics of CLX expressed as the total percentage of drug released (%) from polymer films composed of polymers PCL and PLLA, both HMW and LMW, at a concentration of 50 mg/mL, with CLX loadings of 20%. Abbreviations: CLX: Celecoxib. LMW: Low molecular weight. HMW: High molecular weight. DL: Drug loading. PLLA: poly(L-lactide) (PLLA). PCL: poly(caprolactone). Values represent the mean ± standard deviation (n = 3). Note: In cases where drug release was minimal between sampling points (e.g., HMW-PCL and LMW-PCL), slight decreases in cumulative release were observed due to sample replacement with fresh buffer, which may dilute the drug concentration when no measurable release occurred. This is a known limitation of partial-volume sampling methods
To achieve intermediate CLX release kinetics, PLLA and PCL were blended at varying ratios (Fig. 2). All w/w ratios are expressed relative to a total polymer concentration of 150 mg/mL, which corresponds to the actual formulation conditions used for casting. Increasing PCL content accelerated CLX release, a trend attributed to the formation of polymer-to-polymer interfaces that facilitated water penetration and diffusion of the drug near these interfaces [47,48,49,50]. Due to the limited miscibility of PLLA and PCL, PCL domains aggregate, lowering Tg locally and increasing chain mobility. This facilitates nucleation and improves PLLA’s mechanical properties, including flexibility and toughness [51]. Among tested formulations, both PLLA/PCL blends at 70/80 (w/w) and 80/70 (w/w) showed promising results with intermediate release profiles among the blends studied, releasing 40% of CLX after 3 months without plateauing.
Given the variability in intra-articular (IA) CLX dosing across animal models (1-1.25 mg CLX/kg in mice [52], 0.46 mg CLX/kg/week over 5 weeks in rabbits [53], 0.0292 mg CLX/kg [54] or 1 mg/kg [55] in rats, and approximately 1 mg/kg in sheep [56]), it was assumed that higher doses of CLX could enhance therapeutic effects in both in vitro and in vivo models (e.g., reduced fibrous encapsulation, foreign body reactions (FBR), and modulation of signaling pathways). Hence, CLX concentration in selected formulations was increased 2.4-fold (from 12.5 mg/mL to 30 mg/mL), achieving a drug loading (DL) of 16.6%. This increase aimed to enhance the total amount of drug incorporated into the coating, bringing it closer to the IA doses previously reported in the ovine model. The PLLA/PCL blend at an 80/70 (w/w) ratio was ultimately selected for its balanced release profile (Fig. 2).
Fig. 2
Release kinetics of CLX expressed as the total percentage of drug released (%) from PLLA/PCL blends prepared at 150 mg/mL at different (w/w) ratios (ranging from 50/100 to 90/60) with a CLX loading of 16.6%. Abbreviations: CLX: Celecoxib. PLLA: poly(L-lactide). PCL: poly(caprolactone). (w/w): weight-to-weight ratio. Values represent the mean ± standard deviation (n = 3)
These results led to two emerging candidates for sustained CLX release: HMW-PLLA with 20% DL, which provided a very slow release (< 25% CLX released after 6 months), and PLLA/PCL 80/70 (w/w) with 16.67% DL, which achieved ~ 40% release in 3 months. Both formulations exhibited polymer-drug interactions based on thermal analysis (reductions in Tg, Tm, and Tcc) (Supplementary Fig. 3A) and successfully incorporated CLX in a molecularly dispersed form, as confirmed by the absence of CLX crystalline peaks in XRD and the disappearance of its melting endotherm peak in DSC thermograms (Supplementary Figs. 3A–B), both indicative of an amorphous drug state within the polymer matrix. However, only the PLLA/PCL 80/70 (w/w) blend met the required release profile and progressed to further studies.
DEX-loaded polymer filmsTo effectively address post-surgical acute inflammation, DEX was selected for its potent and pleiotropic anti-inflammatory activity and established clinical use. The goal was to achieve a rapid and controlled release over a 1 to 4-week period by incorporating DEX into an external polymer layer made of PLGA. A high molecular weight (MW) (24–38 kDa) was initially explored, but presented limitations due to its characteristic biphasic release profile (Supplementary Fig. 4) —marked by an initial burst from surface-associated drug, followed by a less predictable sustained release phase driven by bulk polymer hydrolysis [57, 58]. The degradation rate of PLGA decreases with increasing lactide content; however, the 50:50 lactide: glycolide ratio used in this study represents the fastest-degrading composition due to its high hydrophilicity and low crystallinity [59].
Given the limitations of PLGA, particularly its biphasic release profile, an alternative amphiphilic matrix—poly(lactic acid)-poly(ethylene glycol) di-block copolymer (PLA-PEG) (PEG = 5,000 Da)—was evaluated. Di-block copolymers like PLA-PEG can influence polymer-drug interactions differently than blends of individual polymers [60, 61] due to the covalent linkage between the blocks, which creates a more defined and stable microphase separation compared to physical blends [62]. This structural organization can alter drug distribution and water accessibility, ultimately affecting drug release kinetics and solubility. For these reasons, di-block copolymers have been widely used in drug delivery systems [63]. As shown in Fig. 3, PLA-PEG enabled a sustained and more complete release of DEX for approximately one week at a polymer concentration of 200 mg/mL. This accelerated release was likely due to PEG-induced aqueous swelling and the proximity of DEX within the matrix. However, the slow degradation rate of the PLA block could prolong the integrity of the top layer, acting as a persistent barrier over the bottom CLX-releasing layer and thereby limiting CLX diffusion to the release medium over time. To improve control over the release profile and mitigate the limitations associated with PLGA’s biphasic behavior, the effect of PLGA molecular weight (MW) on DEX release was investigated. In addition to the high-MW PLGA used initially, low-MW (LMW) variants (8 kDa) were tested both as standalone matrices and in blends with PLA-PEG (Fig. 3).
Blends of PLA-PEG and LMW-PLGA 75/25 (w/w) showed release profiles similar to pure PLA-PEG, but a 50/50 (w/w) blend extended the sustained DEX release to two weeks (Fig. 3). Increasing the relative amount of LMW-PLGA further enhanced this effect. Optimization studies with different PLA-PEG/LMW-PLGA ratios confirmed that higher LMW-PLGA content produced a sustained release within the desired 1–4-week window. Notably, pure LMW-PLGA exhibited a similar release profile, making it a practical alternative to PLA-PEG. Its selection not only met the therapeutic release target but also simplified the formulation by avoiding polymer blends.
Fig. 3
Release kinetics of DEX expressed as the total percentage of drug released (%) from pure PLA-PEG, pure LMW-PLGA, and PLA-PEG/PLGA blend films. LMW-PLGA was incorporated to the PLA-PEG matrix with different PLA-PEG/PLGA ratios ranging from 25/75 to 75/25 (w/w). Films were prepared at 200 mg/mL with constant drug loadings of 2.44%. Abbreviations: DEX: Dexamethasone. PLA-PEG: poly(lactic acid)-poly(ethylene glycol) di-block co-polymer. PLGA: Poly(lactic-co-glycolic) acid. LMW: Low molecular weight. Values represent the mean ± standard deviation (n = 3)
DSC and XRD analyses were performed to confirm the physical state of the incorporated drugs. For both DEX and CLX formulations, the disappearance of their characteristic melting peaks in DSC thermograms and the absence of crystalline reflections in XRD patterns confirmed that the drugs were molecularly dispersed within the polymer matrices (Supplementary Figs. 3A and B, and 5A and B). For CLX-loaded systems, thermal events and crystallinity indicated good miscibility and a predominantly amorphous dispersion of the drug. Slight reductions in Tg and Tm—particularly in PLLA-based formulations—suggested plasticizing effects and increased polymer chain mobility [47, 64,65,66]. These interactions are consistent with the initial diffusion-driven release observed in PLLA and PLLA/PCL blends. Notably, no direct correlation was found between polymer crystallinity and release kinetics, supporting the idea that other factors—such as porosity and polymer-drug interactions—played a more dominant role [67]. In the case of DEX, minor increases in crystallinity were observed in PEG-containing systems, suggesting weak drug-polymer interactions that may facilitate water uptake and diffusion. Conversely, PLGA and LMW-PLGA matrices remained largely amorphous, aligning with their more sustained release behavior. A full list of thermal parameters (Tg, Tm, Xc) is provided in Supplementary Tables 1 and 2.
This comprehensive screening identified PLLA/PCL (80/70, w/w) with 16.6% DL as the most promising formulation for sustained CLX release, achieving higher drug incorporation while avoiding the plateau effect observed with pure PLLA. For DEX release, both PLA-PEG and LMW-PLGA (8 kDa, 50:50 LA: GA), each with a DL of 2.44%, achieved controlled release within the target 1–4 week window, with LMW-PLGA providing a slightly more sustained release profile and the advantage of a simpler formulation.
Engineering a bilayer drug-releasing polymer coating for the sequential release of DEX and CLXTo enable the simultaneous release of CLX and DEX from the non-biodegradable PCU prosthesis (~ 0.7 × 0.7 × 0.3 cm), a bilayer polymeric coating was developed. The system consists of a first layer releasing CLX to be cast on top of the prosthesis, and a second layer releasing DEX, providing a shorter drug release. Polymer matrices previously identified as optimal candidates (see section Design and optimization of drug-loaded polymer films) were selected for testing in this configuration. While solvent casting was effective for coating one surface, dip coating was chosen for full-implant coverage due to its simplicity, versatility, and compatibility with multilayer deposition [18]. This section explores key aspects of bilayer development, including the impact of solvent interactions between layers and the selection of one optimized polymer matrix for each drug for final implementation.
Given that the DEX layer is prepared using acetone, the potential effect of solvent exposure on the underlying CLX-loaded layer was evaluated. Since acetone can partially disrupt polymer-drug interactions, this investigation was crucial for ensuring stability and consistency in release kinetics. This was explored by DSC, XRD, and drug release evaluation— detailed in Supplementary Figs. 6 and 7. When acetone was applied to PLLA/PCL films, a slight acceleration in CLX release was noted, yet the overall release kinetics remained consistent. However, for pure PLLA, acetone significantly altered the release profile, accelerating CLX release from approximately 10% to over 60% by week 21 (Supplementary Fig. 7).
Evaluation of drug releaseThe release profiles of DEX from the two polymer matrices—PLA-PEG and LMW-PLGA— were evaluated while maintaining a constant CLX-releasing bottom layer composed of the PLLA/PCL blend. PCU implants were coated using a dip-coating approach, where CLX was first incorporated into the PLLA/PCL film using DCM, and DEX was then incorporated into either PLA-PEG or LMW-PLGA using acetone. The main distinction between these bilayer systems lies in the choice of the top-layer polymer, which influenced not only DEX release but also indirectly modulated CLX release kinetics. For clarity, these systems are referred to as Prototype PLA-PEG and Prototype PLGA (Fig. 4A). Together, they provide valuable design insights for dual-drug delivery strategies targeting both acute and chronic phases of inflammation.
Despite the identical CLX-releasing layer, the release profiles of CLX varied depending on the top-layer polymer (Fig. 4B). When LMW-PLGA was used as the top layer, CLX release was faster, likely because the lower molecular weight and higher hydrophilicity of the PLGA facilitated water uptake and swelling, which enhanced buffer infiltration into the underlying CLX-loaded layer. Although both LMW-PLGA and PLA-PEG are capable of water uptake and swelling, the impact of this property on drug release depends on the system architecture. In single-layer matrices with embedded DEX, swelling enhances release directly by facilitating diffusion, thus PLA-PEG showed the fastest release of DEX. In contrast, in the bilayer system, the top-layer swelling modulates access to the underlying drug reservoir, making both the permeability and molecular weight of the top layer critical to the overall release kinetics. As a result, LMW-PLGA as the top layer led to faster release of CLX from the bottom layer. Furthermore, the slower-degrading PLA-PEG formed a more persistent barrier, delaying access of the release medium to the CLX layer and thus slowing its diffusion. This interpretation aligns with previous reports describing swelling-mediated release mechanisms in low-MW polymers [44].
Fig. 4
Schematic representation of the two final prototypes of bilayer polymer coatings (A). Sequential release of DEX and CLX from bilayer polymer coatings composed of a first polymer coating of PLLA/PCL, prepared at 150 mg/mL at 80/70 (w/w) with CLX loading of 16.6%; and a second polymer coating of either PLA-PEG or LMW-PLGA, both prepared at 200 mg/mL with DEX loading of 2.44% (B). Abbreviations: PCU: Polycarbonate urethane. CLX: Celecoxib. DEX: Dexamethasone. PLA-PEG: poly(lactic acid)-poly(ethylene glycol) di-block co-polymer. PLGA: Poly(lactic-co-glycolic) acid. PLLA: poly(L-lactide). PCL: poly(caprolactone). LMW: Low molecular weight. Values represent the mean ± standard deviation (n = 3). Both prototypes share the same CLX-releasing bottom layer (PLLA/PCL), while the top layer differs and carries DEX (PLA-PEG or LMW-PLGA)
DEX release profiles were consistent with our prior results, where PLA-PEG exhibited an initial burst followed by a gradual tapering, while LMW-PLGA provided a more sustained and steady release. Both systems achieved complete DEX release within the first few weeks, effectively meeting the goal of addressing acute post-surgical inflammation.
In vitro degradation of PLA-PEG and PLGA bilayer coated implantsAlthough both bilayer prototypes (PLA-PEG and PLGA) achieved the desired release kinetics for DEX and CLX, they exhibited distinct release profiles. Thus, using these prototypes, degradation studies were conducted to better understand the mechanisms behind their different release behaviors and to validate the hypothesis that PLA-PEG degrades more slowly than PLGA. Coated square implants were immersed in PBS supplemented with 1% (w/v) Tween®80, and periodically analyzed for trends in the change of pH and thickness, as well as surface morphology via FESEM.
Over time, Prototype PLA-PEG showed a gradual decrease in thickness, with a pronounced reduction between 4 and 6 months, corresponding to clear structural degradation and coating disintegration (Figs. 5A and 6). By 12 months, the coating was visibly deteriorated, appearing uneven and incomplete, with remarkable signs of degradation and areas where the prosthesis surface was no longer covered. In contrast, Prototype PLGA initially exhibited a thickness increase—likely due to polymer swelling—followed by a steady decrease over the year (Figs. 5A and 7) [68, 69]. The pH monitoring unveiled additional aspects of the degradation behavior not apparent from thickness measurements alone. While Prototype PLA-PEG maintained relatively stable pH values for the first 4 months, a noticeable decline occurred between months 4 and 6, consistent with the delayed onset of degradation observed in FESEM analysis and expected of PLA. In contrast, Prototype PLGA triggered a sharp pH drop by month 2, which then remained low for the rest of the study (Fig. 5B). These trends reflect the faster breakdown of the PLGA matrix and further support the interpretation that the top-layer degradation rate directly influenced buffer access to the CLX-releasing layer.
Fig. 5
Changes in thickness (A) and pH (B) of Prototype PLA-PEG and Prototype PLGA with time. Abbreviations: PLA-PEG: poly(lactic acid)-poly(ethylene glycol) di-block co-polymer. PLLA: poly(L-lactide). PCL: poly(caprolactone). PLGA: Poly(lactic-co-glycolic) acid. µm: micrometers. Values represent the mean ± standard deviation (n ≥ 1)
These findings help explain the differences observed in CLX release profiles. Although both prototypes had the same PLLA/PCL internal layer for CLX release, faster degradation and reduced tortuosity of the PLGA top layer likely allowed earlier buffer access to the underlying CLX matrix, accelerating drug release. In contrast, the slower degradation of PLA-PEG maintained a more effective diffusion barrier for longer. Thickness measurements were affected by sample cutting and imaging variability because of the angle of the image and should be interpreted as relative trends rather than absolute values (Supplementary Fig. 8). In contrast, pH changes provided a more robust and reproducible indicator of degradation, though results may not fully reflect in vivo behavior, where pH buffering and enzymatic processes differ. Nonetheless, prior work suggests that PLGA degradation kinetics in vitro correlate with in vivo molecular weight loss [134].
Fig. 6
FESEM images of the Prototype PLA-PEG at different times of the biodegradation process. Images are divided into pairs per time point, a top image and a bottom image. The top images correspond with the sagittal cut of the bilayer polymer coating and the PCU implant and were obtained using Zeiss EVO analytical FESEM with a magnification of 100X. The thickness of the polymer coating was measured in these top images. The bottom images correspond with the view from above of the bilayer polymer coating sputter coated with iridium and were obtained using Zeiss UltraPlus analytical FESEM with a magnification of 1000X. Abbreviations: FESEM: Field emission scanning electron microscopy. PLA-PEG: poly(lactic acid)-poly(ethylene glycol) di-block co-polymer. µm: micrometers. EHT: Electron high tension. WD: Working distance. Mag: Magnification
Fig. 7
FESEM images of the Prototype PLGA at different times of the biodegradation process. Images are divided into pairs per time point, a top image and a bottom image. The top images correspond with the sagittal cut of the bilayer polymer coating and the PCU implant and were obtained using Zeiss EVO analytical FESEM with a magnification of 100X. The thickness of the polymer coating was measured in these top images. The bottom images correspond with the view from above of the bilayer polymer coating sputter-coated with iridium and were obtained using Zeiss UltraPlus analytical FESEM with a magnification of 1000X. Abbreviations: FESEM: Field emission scanning electron microscopy. PLGA: Poly(lactic-co-glycolic) acid. µm: micrometers. EHT: Electron high tension. WD: Working distance. Mag: Magnification
Evaluation of drug-releasing polymer coating reproducibilityThe reproducibility of the prototypes was assessed by quantifying the total amount of DEX and CLX per unit of area (µg/cm²) across multiple replicates of square-shaped PCU implants. The Prototype PLGA showed consistent DL for both DEX and CLX, indicating a robust and reproducible coating process with controlled dipping cycles and solvent evaporation. In contrast, Prototype PLA-PEG showed slight variability in drug incorporation between replicates, suggesting challenges in achieving uniform deposition. This may be attributed to uneven polymer film formation, variations in drug-polymer interactions, or greater sensitivity to coating conditions on the complex implant surface. These results indicate that Prototype PLGA offers greater reliability and process reproducibility, while Prototype PLA-PEG may require further optimization to ensure consistent DL (Supplementary Fig. 9).
In vitro assessment of sterility, biocompatibility, anti-inflammatory activity, and efficiency of the bilayer drug-releasing polymer-coated PCU implantsPrior to evaluating the immunomodulatory potential of the bilayer-coated implants, a series of in vitro validations were conducted to ensure compatibility with biological assays. Drug release studies confirmed that both DEX and CLX were released in concentrations exceeding their respective effective thresholds (10 µM for DEX, 25 µM for CLX, based on literature [26]) at key time points—3 h, 3 days, 1 week, 2 weeks, and 4 weeks (Table 1). These intervals were strategically selected to capture distinct drug activity phases: early DEX-driven effects, overlapping DEX and CLX activity, and later CLX-dominant responses.
The release buffer also met sterility criteria, with endotoxin levels remaining below 0.125 EU/mL (Supplementary Fig. 10), validating their suitability for macrophage-based assays. Moreover, no cytotoxic effects were observed in HMDMs exposed to release media from either prototype at any time point, indicating good biocompatibility of both the drugs released and the polymer degradation byproducts (Supplementary Fig. 11). These findings are consistent with previous reports describing the non-toxic nature of polyester degradation products and further support the safety of the materials used [70]. With drug release, sterility, and cytocompatibility confirmed, the subsequent experiments focused on assessing the bioactivity of the released drugs by evaluating their capacity to modulate cytokine secretion in M1-like (classically activated with LPS + IFNγ) pro-inflammatory macrophages.
In vitro evaluation of Immunomodulatory activityThe evaluation of cytokine secretion aimed to confirm that the drugs released from the bilayer polymer coatings retained their immunomodulatory activity. Specifically, it was sought to verify that DEX and CLX, when released at their respective time points, could suppress pro-inflammatory cytokine release from macrophages, events associated with acute and chronic inflammation. This assessment was essential to validate the therapeutic functionality of the coating system in an inflammatory environment. Cytokine secretion by M1-like macrophages was measured following exposure to release media collected from the implant incubations at defined time points. The three key pro-inflammatory cytokines selected were TNF-α (a central mediator of acute inflammation [71,72,
Comments (0)