Materials
3-Aminopropyldimethylethoxysilane (S00750-5 g) was purchased from Fluorochem (UK). Mouse monoclonal anti-CD9 antibody (C-4) (sc-13118), mouse monoclonal anti-TSG101 antibody (sc-7964), were purchased from Santa Cruz Biotechnology (USA). Mouse monoclonal anti-bacterial outer membrane protein-A antibody (OMPA11-M), rabbit polyclonal anti-bacterial outer membrane protein-C antibody (BS-20213R), mouse monoclonal anti-bacterial E. coli elongation factor TU antibody (LS-C128699-100), rabbit polyclonal anti-bacterial flagellin antibody (316137), were purchased from Anawa CliniSciences Group (Switzerland). Rabbit polyclonal anti-bacterial surface layer protein antibody (AA 301–400) was purchased from Antibodies Online (USA). Polyclonal goat anti-mouse immunoglobulins/HRP secondary antibodies (P0447) were purchased from Dako (Denmark) and polyclonal goat anti-rabbit immunoglobulins/HRP secondary antibodies (ab97051) were purchased from abcam (CB2 0AX, United Kingdom). Phosphate buffered saline (PBS, KH2PO4 1 mM, NaCl 155 mM, Na2HPO4.7H2O 3 mM, pH 7.4) was purchased from Thermo Fisher Scientific (USA). Silver/silver chloride paste (60/40), skim milk powder, bovine serum albumin (BSA), tris(hydroxymethyl)aminomethane (Tris) base, polysorbate 20, and ethanol absolute (> 99.8%), were purchased from Sigma–Aldrich (USA). SYLGARD 184 silicon elastomeric kit was purchased from Dow Corning (USA). De-ionized water (DIW) was supplied by a Merck Millipore MilliQ Direct-Q8 (USA). All the chemicals were used as received without further purification.
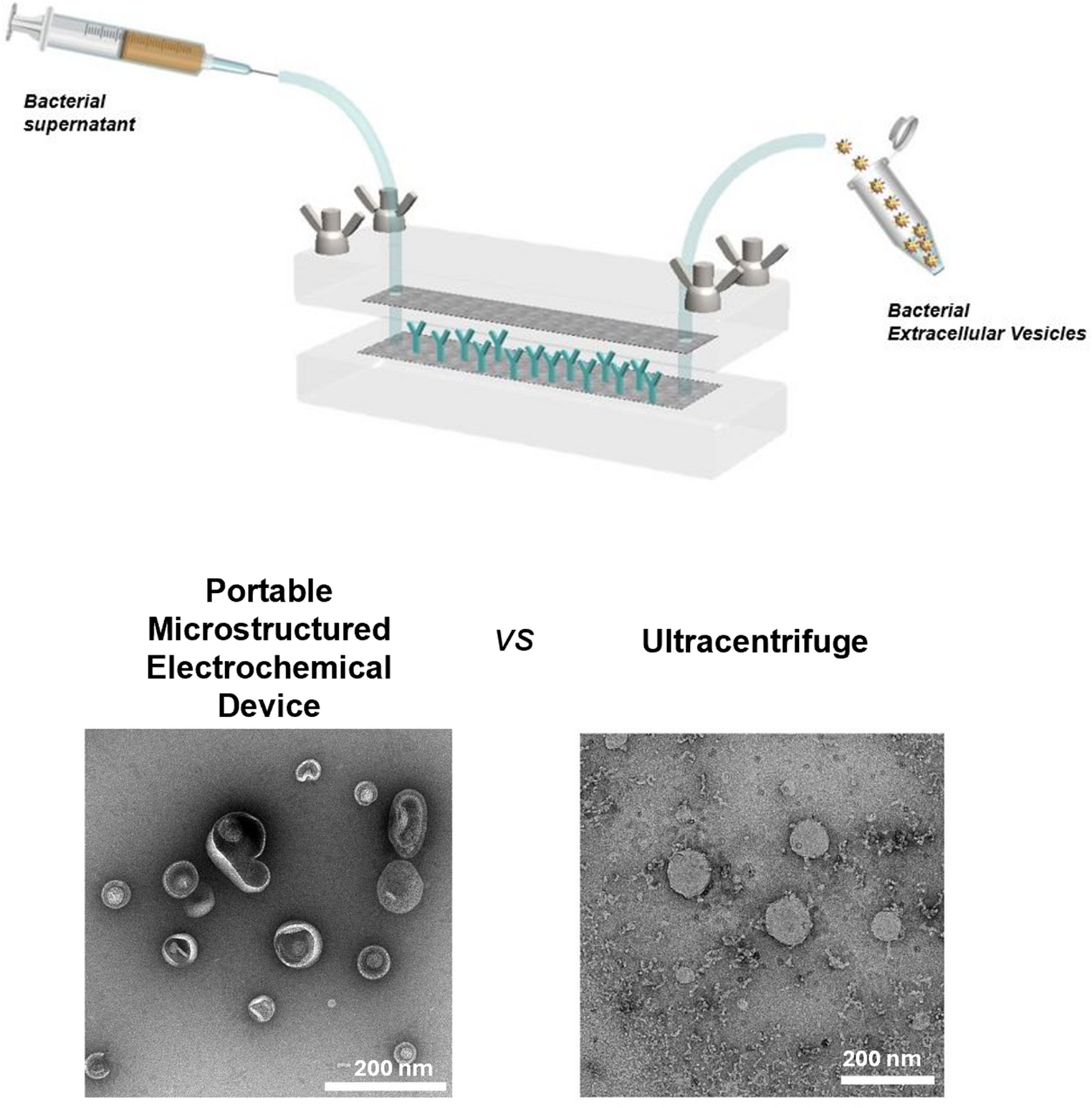
Fabrication of the PMED
The PMED was fabricated as described previously [20]. Briefly, the device consists of a three-dimensional (3D) carbon electrode with a high surface area embedded in a poly(dimethylsiloxane) (PDMS)-based microchannel. The micro-carbon fiber (µCF) electrode is constructed from Freudenberg H23 carbon paper (22 cm × 30 cm, catalog number: 1590042, Freudenberg Performance Materials SE & Co. KG., Germany). The fluidic channel integrates silicon tubing to facilitate the flow of biosamples, which are injected using syringes. The initial step in the device assembly is the fabrication of the microchannels. Two ethanol-washed molds were used for this purpose. Double-sided adhesive carbon tape (8 mm × 20 m, cat. AGG3939, Agar Scientific Ltd, UK) was applied to the molds, and µCF electrodes (Freudenberg H23, 22 cm × 30 cm, 0.15 mm thickness, cat. 1590042, Freudenberg Performance Materials SE & Co. KG, Germany) were positioned on top of the tape. For the reference electrode, a thin 3–4 mm layer of Ag/AgCl paste (60/40% m/m, 901773-50G, Sigma-Aldrich) was applied and then dried in an oven at 80 °C. Silicon tubes (1 mm inner diameter, 3 mm outer diameter, 4.5 cm length, Millipore) were inserted into the counter electrode using needles (100 Sterican ⌀ 0.90 × 50 mm, 20 G×2″, Braun Injekt, Germany). PDMS (50 g) and a curing agent (5 g) were mixed, creating a whitish mixture that was poured onto the molds. The sides of the molds were delimited by microscope slides (26 × 76 × 1 mm, Menzel X50, Thermo Fisher Scientific) covered in aluminum foil. PDMS curing occurred at 80 °C for 1 h. The two semi-channels were then carefully removed, and the electrodes were thoroughly washed with ethanol and deionized water. The resistance of the electrodes and the quality of the contacts were measured using a Digital Multimeter (UNI-T UT61E, Unitrend Technology, China). The conductivity of the working electrode and counter electrode) was approximately 3 Ω cm− 1.
Functionalization of the µCF electrodes
The functionalization of the µCF electrodes has been described in our previous publication [20]. The semi-channels with embedded µCF electrodes were cleaned with 40 mL of ethanol and dried in a vacuum oven at 100 °C for 30 min. A radio frequency (RF) source at 60 W for 1 min, at an oxygen gas pressure of 30 Pa (CY-P2L-B150, Zhengzhou CY Scientific Instrument Co., China), was then used to induce the formation of hydroxyl groups. Silanization was performed via gas-phase chemical adsorption of 300 µL 3-Aminopropyldimethylethoxysilane (APDMES) for 16 h at 100 °C and 0 Pa in a covered glass Petri dish, followed by heating to 105 °C under vacuum for 2 h. The semi-channels were subsequently washed with 50 mL of ethanol and dried under vacuum at 100 °C in a vacuum oven (KVTS11, Salvis, AG., Switzerland).
PMED channel assembly
The assembly and characterization of the PMED channels were performed as described before [20]. The two semi-channels were aligned and combined to form a fluidic channel, then secured between two clear poly(methyl methacrylate) lids (105 mm × 55 mm × 5 mm) using six bolts and nuts. Steel needles were inserted into the PDMS to penetrate the µCF electrodes without touching the fluidic channel and tubing. The fluidic system was flushed with 5 mL of deionized water and 5 mL of PBS. The metal needles were connected to a potentiostat (EmStat3 Blue, PalmSens BV, Netherlands).
Preparation of E. coli samples
The manufacture step involves the bacterial growth via successive unit operations from bacterial cell bank stored at -80 °C up to 400–600 L fermenters stage. The culture medium (200 mL) was collected at the end of the fermentation processes: optical density range 20.00–25.00, measured at 700 nm, (V-730, JASCO, Japan) with > 1.5 × 1010 cells/mL. The culture medium was spun at 10,000 x g for 20 min at a temperature of 4 °C (Allegra X-30R, Beckman Coulter, USA). The supernatant was then collected, filtered with 0.2 μm pore-size filter (567 − 0020 reference, Rapid Flow Nalgene, Thermo Fisher Scientific) and used for subsequent EVs isolation experiments. In addition, 1 mL of sterile supernatant was plated on Tryptone Soy Agar plate (43011 reference, BioMérieux, France) and incubated for 48 h at 37 °C (IF75, Memmert, Germany) to check absence of bacteria cells.
Preparation of Lb. fermentum I3929 samples
Bacteria were grown via successive culture steps from bacterial cell bank stored at -80 °C up to 800-1000 mL lab scale fermenter. The culture medium (250 mL) was collected at the end of the fermentation processes (Multifors, INFORS HT, Switzerland) at optical density values of 3.00-3.50, measured at 700 nm, (NanoDropOne C, Thermo Fisher Scientific) with > 1.0 × 109 cells/mL. The culture medium was spun at 6000 x g for 30 min at 10 °C (Allegra X-30R, Beckman Coulter). The supernatant was then collected, filtered with 0.2 μm pore size filters (Supor EKV membrane in mini Kleenpak capsule, KA02EKVP2S reference, PALL, USA) and used for subsequent EVs isolation experiments. The supernatant was also tested for sterility as mentioned above.
General protocol for isolation by UC
For both bacterial strains, 25 mL of supernatant were centrifuged at 110,000 x g and 4 °C for 60 min using an Optima XE-90 ultracentrifuge (Beckman Coulter), equipped with a Type 45 Ti Fixed-Angle Titanium Rotor. The resulting pellet was resuspended in 25 mL PBS, pooled and subjected to a second UC run under the same conditions. The final pellet was resuspended in 1 mL of PBS and stored at 4 °C. After characterization, the aliquots were stored at -20 °C.
Isolation of bacterial EVs from E. coli and Lb. fermentum I3929 samples with the PMED
For the isolation of EVs from E. coli and Lb. fermentum I3929 with the device, the electrodes were coated with 20 µg of the antibody, (100 µg/mL in 200 µL PBS), using a 1-mL syringe. An empty 1-mL syringe was used at the other end of the channel to collect the solution. The antibody solution was incubated for 1 h at room temperature and transferred between the two syringes every 10 min, facilitating mixing and re-distribution of the antibodies. This dynamic flow approach helps ensuring even deposition of the antibodies on the carbon surface.
After incubation, the non-adsorbed antibody solution was removed using a 1 mL syringe, followed by washing with 5 mL of PBS to remove remaining non-bound antibodies. Then, 25 mL of E. coli supernatant or of Lb. fermentum supernatant were injected into the fluidic channel at a rate of 0.3- 1 mL/s and recirculated ten times. The remaining solution was then withdrawn, and 40 mL of PBS were injected at 0.3 mL/s to remove contaminants. For the release step, 1 mL of PBS was injected and then withdrawn from the device. Before the electrochemical release, a 1-mL syringe filled with PBS was connected to one end of the device, and an empty 1 mL-syringe was connected to the other end. The device’s needles were connected to a potentiostat, and a potential of − 1.5 V was applied to the working electrode for 45 s while the solution was cycled back and forth through the channel for 10 times. This process was repeated with the electrical poles reversed. As a negative control, only PBS solution (no bacteria or additives) was processed using the PMED device, and no particles were detected by NTA. The same experimental conditions described for the experiment with E. coli supernatant, were applied.
Preparation of E. coli-contaminated urine sample and isolation of bacterial EVs with the PMED
Urine (Human Urine Male, BioIVT, LOT# NMN563177, UK) biosample was stored at − 80 °C. The initial concentration of E. coli culture media was 107 CFU/mL. Given that counts as low as 102 CFU/mL in urine can indicate an infection [24], a series of 1:10 serial dilutions was performed to progressively reduce the bacterial concentration. First, 1 mL of the 107 CFU/mL culture was transferred into 9 mL of sterile PBS. The mixture was then thoroughly mixed. From this diluted sample, 1 mL was transferred into another tube containing 9 mL of PBS. This process was repeated until the bacterial concentration reached 104 CFU/mL. To achieve a final concentration of 102 CFU/mL in 25 mL of urine, 0.25 mL of the 104 CFU/mL dilution was added directly to the urine sample and mixed thoroughly. Finally, the resulting 25 mL of contaminated urine sample were introduced into the device channels without any pre-purification step, and EVs were isolated, following the protocol described in Section “Isolation of bacterial EVs from E. coli and Lb. fermentum I3929 samples with the PMED”. To exclude the presence of EVs from Gram-negative bacteria and eukaryotic cells, three devices were modified with anti-OmpA and tested with urine only. Following the application of standard electrical potential, no particles were detected by NTA.
Atomic force microscopy (AFM)
AFM topography imaging was conducted in tapping mode under ambient conditions using an AFM system from Horiba, France. Si cantilevers from AppNano (USA) with a spring constant of 2.7–44 N/m and a resonance frequency of 68–290 kHz were employed. Imaging was performed at a scan rate of 0.5–5 Hz. AFM topography images were analyzed using the open-source software Gwyddion (Version 2.49, Czech Republic) [25]. The root mean square (RMS) was calculated based on Eq. 1:
$$\:RMS=\:\sqrt\sum\:_^_^}$$
(1)
Where:
N = number of data points.
j = index.
\(\:_=\:_-\:\stackrel\), where:
\(\:\stackrel=\:\)mean height of the surface [25].
Preparation of the antibody, vesicles and antibody-vesicle complex solution on mica substrate for AFM imaging
The mica surface underwent the same functionalization treatment as the carbon fibers, which included oxygen plasma and silanization [20]. To prepare the OmpA antibody solution, 20 µL of the antibody solution were drop-cast directly from the original batch onto the mica surface and then dried at 40 °C for 48 h. For the E. coli EVs, 25 mL of the original supernatant were processed through UC (Section “General protocol for isolation by UC”). The resulting pellet was resuspended in 1 mL of PBS. Subsequently, 20 µL of this EV solution were drop-cast onto the mica surface and dried at 40 °C overnight. The antibody-vesicle complexes were prepared by gently mixing post-UC EVs with OmpA antibodies from their original stock solution at a volume ratio of 1:2 (EVs:OmpA), resulting in a final volume of 150 µL. The mixture was incubated at room temperature for 30 min to ensure efficient binding of the antibodies to the OmpA molecules on the EV surfaces. Following incubation, 20 µL of resulting solution were then drop-cast directly onto the mica surface and dried at 40 °C overnight.
Preparation of the antibody-vesicle complexes on device fibers for AFM imaging
To prepare the OmpA antibody-EV complex on the device fibers, the isolation procedure described in Section “Isolation of bacterial EVs from E. coli and Lb. fermentum I3929 samples with the PMED” was followed without applying electrical voltage, as the objective was to image vesicle binding to the antibodies rather than their release. After introducing 25 mL of supernatant into the antibody-modified channels and performing a PBS wash, the fibers—now containing the antibody-vesicle complexes—were carefully removed from the carbon tape using tweezers, separated, and placed on a glass substrate. The substrate was then incubated at 40 °C overnight, covered with a cup to prevent dust contamination. Once dried, the fibers were transferred onto an atomically flat mica surface and placed under the AFM microscope for imaging.
Nanoparticle tracking analysis (NTA)
NTA was used to estimate the particle concentration and the size profile of the isolated bacterial EVs. For the measurement, a ZetaView® PMX 120-Z instrument (Particle Metrix GmbH, Germany), featuring a 405-nm laser source and a CMOS camera, was used. The NTA device was calibrated with a 1:500,000 dilution of silica beads. To measure our samples, the shutter was set to 150, sensitivity to 85, and the frame rate to 30. Data were analyzed with the ZetaView software version 8.04.04 SP2, (Particle Metrix GmbH), applying a bin class width of 5 nm, a minimum brightness of 25, a minimum area of 5, and a maximum area of 1000, A 1:50 (v/v) dilution of the EV samples was inserted using a syringe and measured for each sample in triplicates. The Savitzky-Golay filter (Points of Window 10) was used for smoothing data by fitting successive subsets of adjacent data points with a second-degree polynomial function. The NTA plots show the mean particle size and concentration, derived from 3 independent experiments, conducted each in triplicates.
Dynamic light scattering (DLS)
The hydrodynamic diameter (nm) and zeta-potential (mV) of the vesicles were measured on a Malvern Zetasizer Advance Pro 722 instrument (Malvern Panalytical, UK) operating at a scattering angle of 179° and at a temperature of 25 °C. The hydrodynamic diameters were determined based on light scattering intensity, with samples diluted 1:10 (v/v) PBS in nanopure water, and measured in 70 µL micro UV-cuvettes (Brand GmbH + CO. KG., Germany). The zeta-potentials were performed via laser Doppler anemometry, with samples diluted with 10% (v/v) PBS in nanopure water and analyzed in 800 µL Folded Capillary Zeta Cell cuvettes (Malvern Panalytical GmbH, Germany).
Transmission electron microscopy (TEM)
TEM was used to image EVs. To this end, 300-mesh copper grids with ultrathin carbon support film were negatively glow-discharged in a Pelco EasiGlow discharge system (Ted Pella Inc., USA) at 25 mA for 30 s at a pressure of 38 Pa. Subsequently, 4 µL of the EV solution were deposited onto the grids for a 30 s absorption period. The grids were treated with a uranyl acetate staining solution and washed with distilled water. Excess liquid was carefully removed using filter paper, and the grids were dried under ambient conditions. The samples were imaged with a Tecnai F20 field emission gun microscope equipped with a combination of a CCD camera (ORIUS SC200 2 K, Gatan, USA) and a direct electron detector (Falcon II 4 K, Thermo Fisher Scientific) at an acceleration voltage of 120 kV. The micrographs were acquired in the bright field mode.
Cryo transmission electron microscopy (Cryo-TEM)
To prepare samples for cryo-TEM, lacey carbon EM grids, (Au 300 mesh, 150 μm, Byolist Scientific, USA), were glow-discharged in Pelco EasiGlow system (Ted Pella Inc) at 25 mA for 30 s at a pressure of 38 Pa. Subsequently, 3.4 µL of the EV solution were applied onto the carbon side of EM grid, which was then blotted for 2.0 s and plunge-frozen into the precooled liquid ethane: propane mixture with Vitrobot Mark IV (Thermo Fisher Scientific). This procedure applied a thin layer of amorphous ice to the samples, preserving them in their native state and shielding them from potential radiation damage. In order to obtain a good ice thickness, the blotting time was set to 2.5 s, the blotting force to 0, and the temperature to 22 °C. The cryo-TEM images were acquired at Titan Krios FEG (Thermo Fisher Scientific) at 300 kV using a direct electron detector (Falcon III 4k × 4k) (Thermo Fisher Scientific), which works in tandem with a Ceta 16 M 4k × 4k CMOS detector (Thermo Fisher Scientific) and K2 (Gatan, USA) with the Quantum LS energy filter (Gatan).
Determination of bacterial EV protein concentration
The total protein content of bacterial EV samples was measured with the Micro BCA Protein Assay Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. The preparation of BSA standards is described in Table S1. Bacterial EVs samples were prepared by diluting 5 µL of sample with nanopore water to a volume of 150 µL. After addition of 150 µL Micro BCA reagent (25:24:1 (v/v), reagent A:B:C) per well, the plate was incubated at 37 °C for 90 min in the dark. The absorbance was measured at 562 nm using the Tecan Infinite M200 plate reader (Tecan, Switzerland). Protein concentration was determined by accounting for the dilution of the EV sample.
Western blotting
Sample buffer (0.25 M Tris, 10% w/v SDS, 30% v/v glycerol, 0.02% w/v bromophenol blue, 5% v/v β-mercaptoethanol) was added 5:1 (v/v) to the bacterial EV samples to denature proteins and promote a sufficient flow on the gel. The samples were then heated for 5 min at 95 °C. Following this, the samples and the size marker were loaded on a 12% SDS polyacrylamide gel (total final volume loaded in each well was 15 µL). The protein samples (1–7 µg) were resolved at 80 V for approximately 1.5 h. The transfer to polyvinylidene fluoride membranes was performed with a Trans-Blot® Turbo™ system at 25 V for 10 min (Bio-Rad, USA). Blocking was conducted for 2 h using Tris-buffered saline (20 mM Tris, 150 mM NaCl) with 0.1% (v/v) polysorbate 20 (TBS-T) and 5% (w/v) skim milk powder for OmpA, OmpC, flagellin, TSG101 and CD9. Instead, blocking was conducted for 2 h using 8% (w/v) BSA in tris-buffered saline (TBST) for Elongation TU, while 15% (w/v) BSA in TBS-T was used for ENO-1 and S-layer. These conditions were optimized to minimize non-specific antibody interactions while maintaining effective target binding. Following this incubation time, the blocking solution was discarded, and the membranes were incubated with primary mouse monoclonal anti-bacterial antibodies including OmpA and Elongation TU, or with primary rabbit polyclonal anti-bacterial antibodies including OmpC, flagellin, S-layer and ENO1 in TBS-T (1:200 v/v). Primary antibodies were incubated overnight at 4 °C. After three washes with TBS-T (20 min, room temperature), secondary horseradish peroxidase (HRP)-conjugated antibodies in TBS-T were added for 4 h at 4 °C. Antibody concentrations (v/v) are provided in Table S2. Membranes were incubated with Western Blotting Luminol reagent (Santa Cruz Biotechnology, USA) according to the manufacturer’s protocol and imaged using a ChemiDoc MP Imaging System (Bio-Rad).
Nano flow cytometry (NanoFCM)
Bacterial EVs were labeled with 5 µM CellTrace™ Far Red dye (Thermo Fisher Scientific) in PBS, following a previously described protocol [26]. Briefly, samples were incubated overnight at 4 °C with the dye, followed by 15 min at 37 °C to promote acetate hydrolysis of the dye. Excess dye was removed by dialysis against PBS overnight at 4 °C using the Pur-A-Lyzer™ Mini 6000 Dialysis Kit (Sigma-Aldrich).
Samples were analyzed using a Flow NanoAnalyzer N30 instrument (NanoFCM Co., Ltd., UK). Light scattering and fluorescence from individual particles were collected for 1 min across three channels: 488/10, 525/40, and 670/30 using single-photon counting avalanche photodiode (SPCM APD) detectors. Channel alignment was performed with fluorescent 250 nm silica QC beads and particle size was determined according to standard operating procedures by generating calibration curves using the S16M-Exo silica nanosphere cocktail (both NanoFCM Co., Ltd.), which includes four distinct silica particle populations with diameters of 68 nm, 91 nm, 113 nm and 155 nm. Samples were measured with lasers set to 10/50 mW (488 nm) and 20/100 mW (638 nm) with a 10% side scatter decay. The sampling pressure was maintained at 1.0 kPa throughout the measurement. Before measurement, the samples were diluted in PBS to ensure a final event count ranging from 500 to 3200. The data was processed using the NanoFCM Professional Suite (version 2.3, NanoFCM Co., Ltd.) and the FlowJo software (version 10.9.0, BD Biosciences, USA). Experimental data are shown as a result of 3 independent experiments, conducted each in triplicates.
Proteomic analysis—EVs proteome analysis
Two Lb fermentum EV samples (post-device) were dissolved in 1% (w/v) sodium deoxycholate in 50 mM ammonium bicarbonate buffer (pH 8), reduced with 1 mM dithiothreitol, and alkylated using 5.5 mM iodoacetamide for 10 min at room temperature. Proteins were in-solution digested with trypsin (Promega, USA). Sodium deoxycholate was precipitated using 50% TFA (trifluoroacetic acid), and tryptic peptides were purified by STAGE tips prior to LC–MS/MS measurements. These were performed on a QExactive HF-X mass spectrometer (Thermo Fisher Scientific) coupled to an EasyLC 1000 nanoflow-HPLC (Thermo Fisher Scientific). Peptides were separated on a fused silica HPLC-column tip (I.D. 75 μm, New Objective, self-packed with ReproSil-Pur 120 C18-AQ, 1.9 μm (Dr. Maisch, Germany) to a length of 20 cm), using a gradient of A (0.1% v/v formic acid in water) and B (0.1% v/v formic acid in 80% v/v acetonitrile in water). The mass spectrometer was operated in data-independent mode. After each survey scan (mass range m/z = 350–1200; resolution: 120,000), 28 DIA scans with an isolation width of 31.4 m/z were performed, covering a precursor range of 350 to 1200 m/z. AGC target value was set to 3 × 106, resolution to 30,000, and normalized collision energy to 27%. Data were analyzed using Spectronaut software version 15.7 (Biognosys, Switzerland) with standard settings (without imputation) in direct DIA mode, using the reference proteomes uniprotkb_Limosilactobacillus fermentum and uniprotkb_Rabbit (UniProt, full length) and common contaminants.
Cell stimulation assays
HEK-Blue™ hTLR2: hkb-htlr2 and HEK-Blue™ hTLR4: hkb-htlr4 (Invivogen, USA) were plated at a density of 5.104 cells / well of a 96 well plate in 100 µL of culture medium (DMEM high glucose glutamax, Gibco, 61965-026 + 10% v/v fetal calf serum, Gibco A52567, 10% fetal bovine serum (FBS), Thermo Fisher Scientific). After 24 h of incubation, the medium was replaced with fresh medium, and various dilutions of EVs isolated from Lb. fermentum culture supernatant were added to the cells. After 24 h of incubation 37 °C under 5% CO2 atmosphere, SEAP activity was revealed using QUANTI-Blue reagent (Invivogen) and a Fluorescence plate reader (SpectraMax M3. Molecular Devices, USA) according to manufacturer’s instruction. Results were expressed by comparison between the basal Optical Density (OD) measured in cells supernatant without any treatment and OD obtained after cells stimulation. The NF-kB activity was calculated using Eq. 2
$$\:NF-\kappa\:B\:activity=\frac\text\:\text\text\:\text\text\text\text\text\text\text\text\text\text\:\text\text\text\text\text}\text\:\text\text\:\text\text\text\text\:\text\text\text\text\text\text\text\text\:\text\text\text\text\:\text\text\text\text\text\text}$$
(2)
This ratio was expressed as activity “relative to unstimulated cells”; a ratio equal to 1 corresponding to absence of NF-kB activity.
Statistical analysis
The statistical analysis was conducted using the GraphPad Prism software version 8.0.0, (GraphPad Software, Inc., USA). The data were presented as mean ± SD. p-values were calculated using an unpaired t-test with a two-tailed distribution and unequal variance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.

Comments (0)