Participants
Participants were healthy Chinese adults aged 18 to 50 years, with a body weight between 40 and 100 kg, and a body mass index (BMI) ranging from 18.0 to 28.0 kg/m2. Eligible individuals were required to have no plans for parenthood and to take reliable contraceptive methods during the study and for 6 months after completion. Additionally, a tuberculosis infection T cell spot test result of ≤ 2 times the upper limit of normal was required within 28 days prior to dosing.
Participants were excluded if they recorded any clinically significant abnormalities in physical examination, blood biochemical tests, pancreatic function tests, complete blood counts, clinical urine tests, electrocardiograms, or abdominal ultrasonography; had a history of allergic diseases (including asthma and urticaria); or had a known history of allergies to firsekibart, any of its ingredients, or similar drugs. In addition, individuals were excluded if they had a history of cardiovascular, haematological, hepatic, gastrointestinal, renal, neurological, psychiatric, or metabolic disorders considered clinically significant by the investigator; smoked > 5 cigarettes per day; or had participated in any clinical trial within 3 months prior to dosing.
Study Design
This randomized, double-blinded, single-dose, dose escalation, placebo-controlled, first-in-human, phase 1 trial (ClinicalTrials.gov identifier NCT04337437) was conducted at a single centre in China.
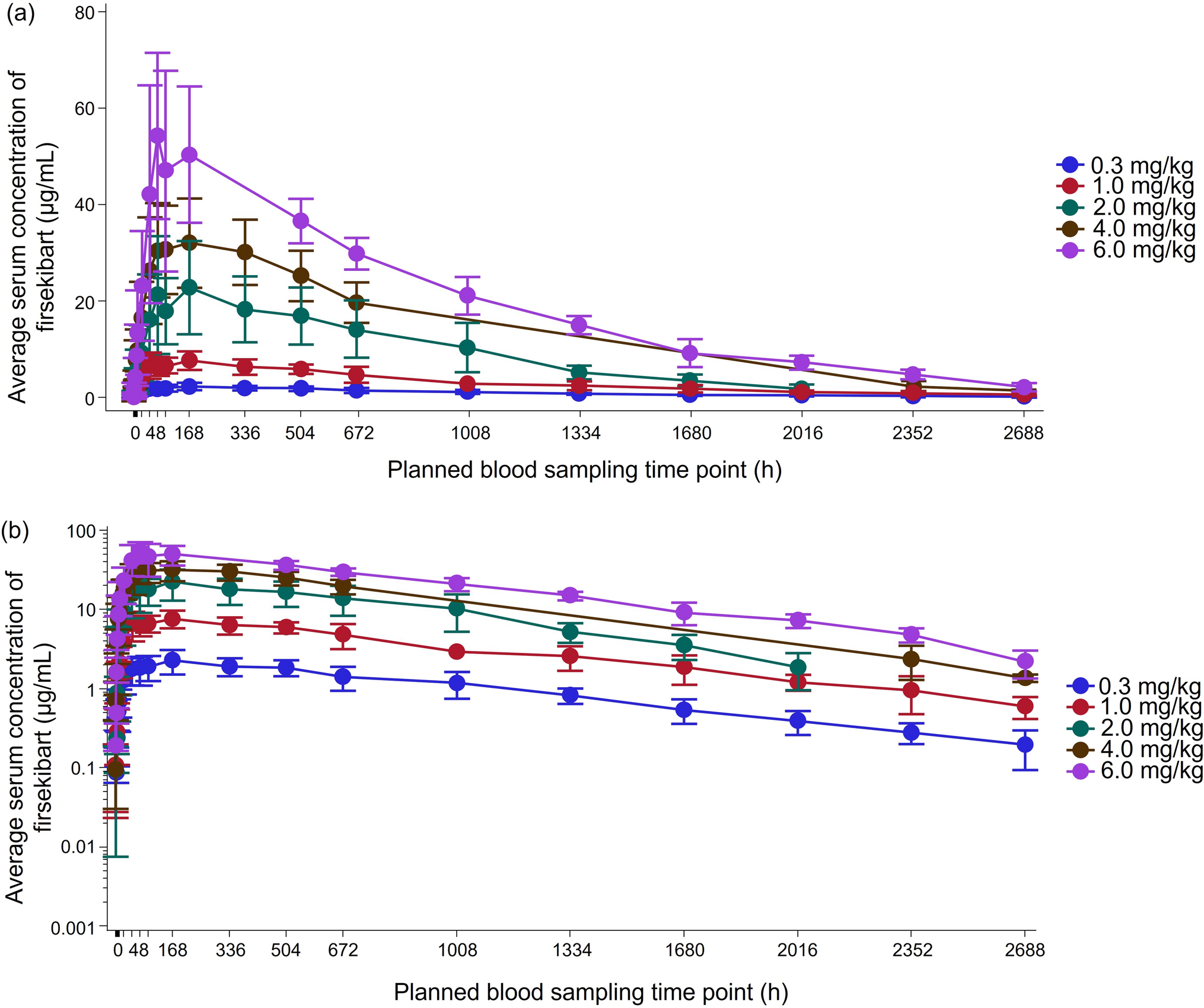
Participants were assigned to five sequential dose-escalated groups: 0.3, 1.0, 2.0, 4.0, and 6.0 mg/kg. Firsekibart was administered as a single dose to each participant, and no subject received multiple doses or participated in more than one dose group. In each group, participants were randomized in a 3:1 ratio to receive firsekibart or placebo, administered as a single subcutaneous injection of equal volume. In the 0.3 mg/kg group, two individuals who were blinded to treatment allocation were the first to enter the study (‘sentinel individuals’) and were randomized 1:1 to receive firsekibart or placebo. To minimise the risk of acute allergic reactions, the remaining participants in the 0.3 mg/kg group were treated 48 h after the sentinel participants. Participants were then assessed by a non-blinded investigator on day 22. If the discontinuation criteria for dose escalation were not met (as detailed in Supplementary Material, Fig. S1), the same process of assignment, treatment, and assessment was repeated for the remaining dose-escalated groups (1.0 to 6.0 mg/kg) consecutively. All participants were observed until day 113 (Supplementary Material, Fig. S2).
The sampling schedule was designed on the basis of the anticipated PK properties of firsekibart based on prior preclinical studies including its long terminal elimination half-life (t1/2; unpublished data). Timepoints (within hours to several days post-dose) were selected to capture absorption and distribution phases, with day 113 included to ensure adequate coverage of the drug’s elimination phase (about 4 half-lives) and to monitor the duration of pharmacodynamic effects and potential development of anti-drug antibodies (ADAs). This extended sampling period was selected to allow for comprehensive assessment of the exposure, immunogenicity, and pharmacodynamic profiles of firsekibart over time.
The minimum recommended starting dose of 0.3 mg/kg was determined on the basis of the starting dose for escalation of canakinumab in healthy individuals (1.1 mg/kg) [9], the predicted minimum effective dose of firsekibart in humans (≤ 0.523 mg/kg), and the anticipated greater cell-based potency of firsekibart compared with canakinumab (unpublished data). The maximum dose of 6 mg/kg was considered appropriate to ensure safety in healthy Chinese adults, based on the maximum escalated dose of canakinumab (900 mg) [5], and the anticipated comparable safety profile of firsekibart with canakinumab.
Ethical Approval
This study was approved by the Ethics Committee at Peking Union Medical College Hospital (approval number HS2019005). The study was conducted in accordance with the local legislation and institutional requirements, and the study was conducted in compliance with the ethical standards established in the 1964 Declaration of Helsinki and its subsequent amendments, with the exception that this trial was retrospectively registered at ClinicalTrials.gov. This trial was prospectively registered on the Chinese Clinical Trial Register (chinadrugtrials.org; registration number CTR20190319) in 2019, prior to first patient enrolment, and subsequently registered on ClinicalTrials.gov in 2020 after study initiation. All participants provided written informed consent before the study.
Objectives
The primary study objective was to evaluate the safety and tolerability of firsekibart as a single-dose, subcutaneous injection in healthy, Chinese adult volunteers. Additional objectives were to evaluate the PK, immunogenicity, and PD of firsekibart in the study population.
Safety Assessments
Safety was assessed throughout the study by monitoring the frequency, duration, severity, relationship with study drug, and outcomes (whether an event led to treatment discontinuation or required medical intervention) of adverse events (AEs) and serious AEs (SAEs). AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0, and the severity of AEs was graded using Common Terminology Criteria Adverse Events (CTCAE) version 5.0. Treatment-emergent adverse events (TEAE) were defined as AEs that exacerbated after administration or novel AEs observed after administration. Other safety measurements included vital signs, physical examinations, laboratory tests, an electrocardiogram, and abdominal B-ultrasonography.
Immunogenicity Assessments
Blood samples for ADA analysis were collected pre-administration (day 0), and on days 8, 15, 29, 57, 85, and 113 post-administration. The incidence of ADAs in serum was evaluated to determine drug immunogenicity. Positive ADA samples were further analysed for titer and neutralising antibody (NAb) status. An electrochemiluminescence (ECL)-based method was employed to detect potential ADAs in serum samples. Additionally, a competitive ligand-binding assay (CLBA) technology, based on an enzyme-linked immunosorbent assay (ELISA) platform, was used to qualitatively detect anti-firsekibart NAbs in the serum.
Pharmacokinetics and Pharmacodynamics Assessments
Blood samples to detect the serum concentration of firsekibart, total serum IL-1β, free IL-1β, interleukin-1 receptor antagonist (IL-1Ra), interleukin-6 (IL-6), and tumour necrosis factor α (TNFα) were collected pre-administration (day 0) at 0.5, 1, 2, 4, 8, and 12 h post-administration, and on days 2, 3, 4, 5, 8, 15, 22, 29, 43, 57, 71, 85, 99, and 113 post-administration. The term “serum concentration of firsekibart” refers to the free drug concentration, as measured using a validated assay; serum concentrations of firsekibart were used for PK evaluation. PK parameters were estimated using a non-compartmental model in Phoenix WinNonlin 8.1 software, including peak serum drug concentration (Cmax), time to reach the peak serum concentration following drug administration (Tmax), area under the concentration–time curve from zero to last measurable concentration (AUC0–t), area under the concentration–time curve from zero to infinity (AUC0–∞), t1/2, terminal rate constant (λz), apparent clearance (CL/F), apparent volume of distribution (Vz/F), and percentage of extrapolated area under the concentration–time curve (AUCextrap%). An ELISA was used to determine the concentration of firsekibart in serum. The lower and upper limits of quantification (LLOQ; ULOQ, respectively) for the serum assay were 25.0 and 2000 ng/mL, respectively.
Total serum IL-1β (sum of IL-1β bound to firsekibart plus free IL-1β) was the primary PD marker. Other exploratory PD markers included serum free IL-1β, IL-1Ra, IL-6, and TNFα. Single molecule immunoassay analysis was used to determine the total IL-1β in serum (total IL-1β: LLOQ 0.800 pg/mL and ULOQ 200 pg/mL; free IL-1β: LLOQ 0.400 pg/mL and ULOQ 200 pg/mL). An ELISA was used to quantify the concentration of cytokines IL-6 and TNFα in human serum (IL-6: LLOQ 0.730 pg/mL and ULOQ 374 pg/mL; TNFα: LLOQ 0.315 pg/mL and ULOQ 162 pg/mL). An ECL assay was used to quantify the concentration of IL-1Ra in serum (LLOQ 22.9 pg/mL and ULOQ 366 pg/mL).
Statistical Analysis
As typical for phase 1 trials, the sample size was not calculated on the basis of statistical considerations. The data from participants receiving placebo in each dose group was merged. Baseline characteristics were assessed from the intention-to-treat (ITT) population, defined as all participants randomized in the trial. Safety analyses were conducted on the safety set (SS), comprising all participants in the ITT population who received firsekibart or placebo. PK and PD evaluations were investigated in the PK set (PKS) and PD set (PDS), respectively, including all participants who received firsekibart, had no important protocol deviations impacting PK/PD results, and had ≥ 1 evaluable PK or PD parameter, respectively. Safety, tolerability, PK, and PD data were summarized with descriptive statistics. The correlation of AUC0–t, AUC0–∞, and Cmax with the administered firsekibart doses was evaluated using a power model based on log-transformed data: ln(PK) = α + β × ln(Dose), where β represents the slope of the regression line. Dose-proportionality was assessed by constructing the 90% confidence interval (CI) for β; if the 90% CI fell entirely within the predefined interval, dose proportionality was concluded. Statistical analysis was performed using SAS 9.4 software.

Comments (0)