Study Design and Oversight
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation Good Clinical Practice (ICH-GCP) guidelines, and the appropriate regulatory requirements in the countries in which the study was conducted. The protocol, its amendments, and informed consent documentation were reviewed and approved by the institutional review board at the site. All participants signed an institutional review board/independent ethics committee-approved informed consent form before any study-specific procedures were performed. This trial is registered with the European Union Clinical Trials Register (EudraCT 2018-004033-33; date of registration, 17 October 2018).
Study Population
Healthy male and female volunteers aged 18–55 years were eligible for the study if they had a body weight of 60.0–100.0 kg and a body mass index of 18.5–29.9 kg/m2. Participants had to be non-smokers or light smokers. Light smokers were defined as those who smoked not more than 5 cigarettes, 1 cigar, or 1 pipe per day. They agreed to abstain from smoking while they were hospitalized in the study institution.
Participants were excluded from the study if they received ustekinumab or its biosimilar before the study; any other monoclonal antibody or fusion protein within 9 months before the administration of the study drug; or any prescription drug or any over-the-counter drug within 2 weeks (or less than 5× the half-life of the drug, whichever was longer). Other exclusion criteria included a history or complication of (1) cardiac, hepatic, renal, gastrointestinal, respiratory, neurological, central nervous, mental, or hematological function disorder; (2) malignancy of any organ system; and (3) allergic symptom such as latex hypersensitivity, asthma, drug-induced rash, or urticaria. Female participants of childbearing potential had to have a negative serum pregnancy test result. Female participants of childbearing potential and male participants who had female partners of childbearing potential agreed to use two forms of birth control throughout the study.
Study Treatment
The study consisted of a screening visit (day − 28 to day − 2), a treatment and in-house observation period (day − 1 to day 3), an outpatient observation period of about 15 weeks with 22 visits, and an end-of-study visit on day 112 (week 16). Participants were hospitalized in the study institution from day − 1 to day 3, and the study drug was administered on day 1.
Participants were randomly assigned in a 1:1:1 ratio to receive a single subcutaneous injection (45 mg) of DMB-3115, EU-Stelara, or US-Stelara. Participants were stratified by body weight (< 80 vs. ≥ 80 kg). On day 1, participants fasted for 8 h before the drug administration and 1 h thereafter. Fluid intake was restricted from 1 h before the drug administration until 1 h thereafter. All study drugs were similar in appearance but not identical. Therefore, handling and administration of the study drugs were performed by the independent, unblinded study site personnel not involved in the study assessments.
Outcome Measures
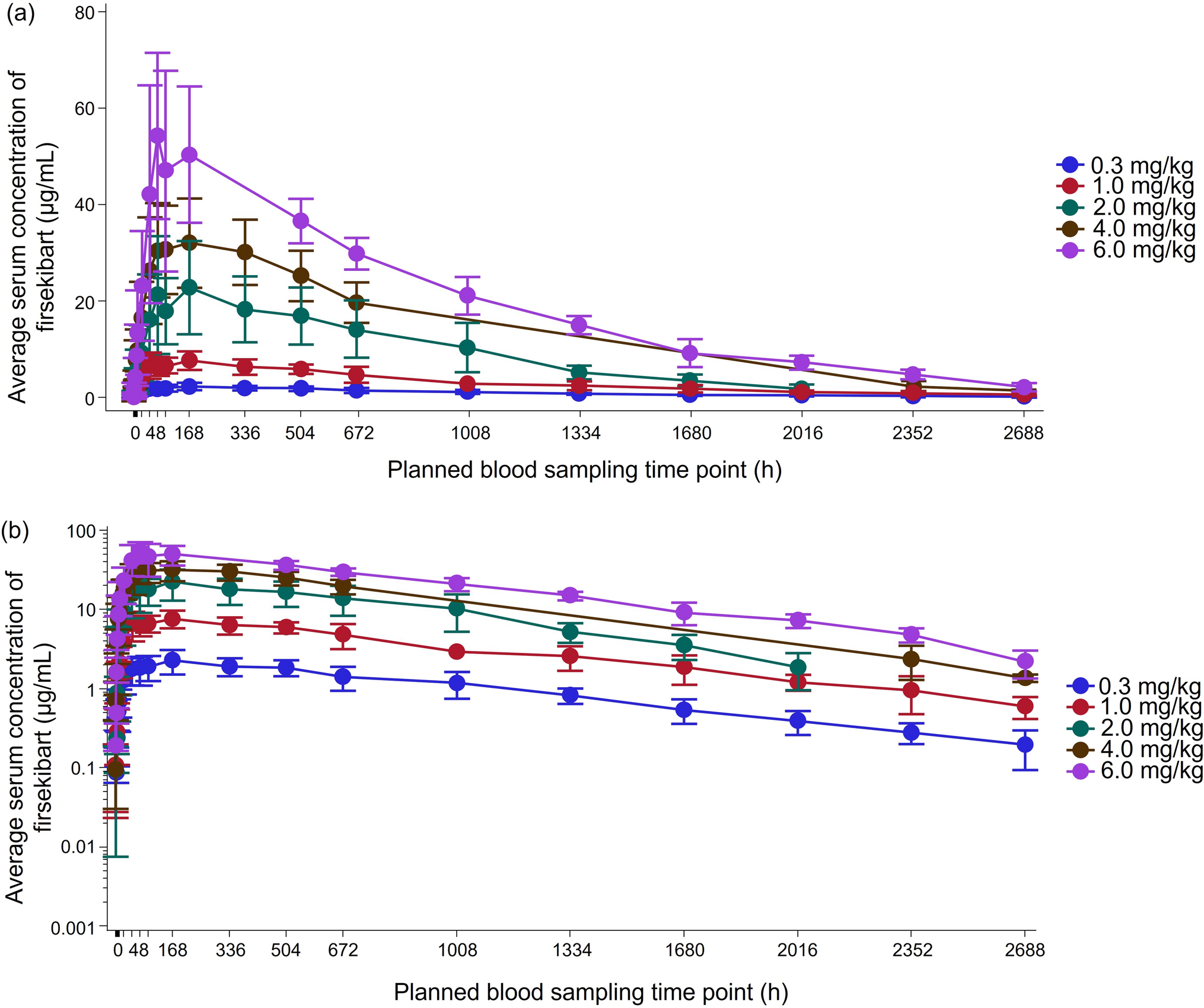
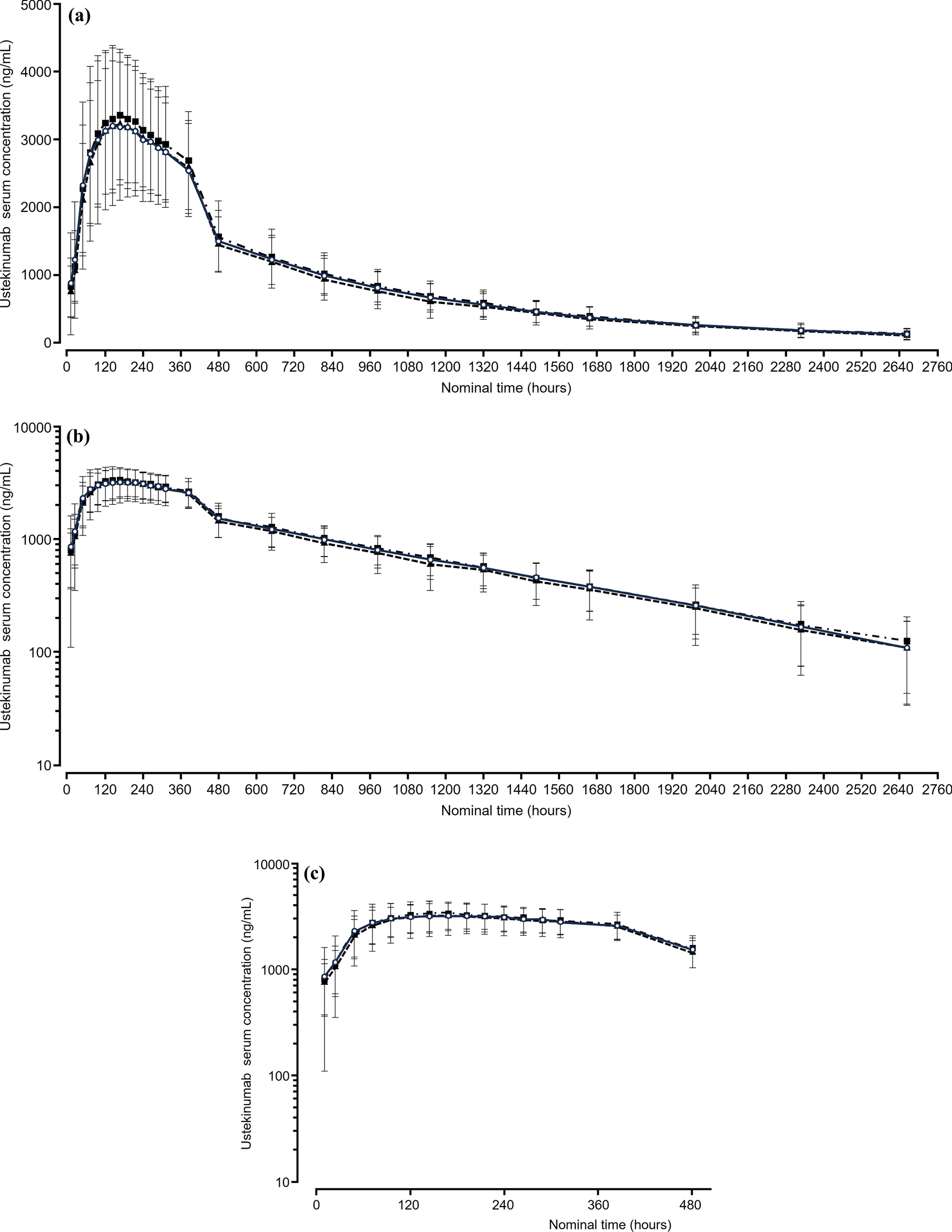
Blood samples for PK analysis were collected on day 1 at pre-dose and 12 h post-dose as well as at 24, 48, 72, 96, 120, and 144 h post-dose, and on days 8, 9, 10, 11, 12, 13, 14, 17, 21, 28, 35, 42, 49, 56, 63, 70, 84, 98, and 112. PK, anti-drug antibody (ADA), and neutralizing antibody (NAb) assays were developed, validated, and performed by Synexa Life Sciences Ltd. (Cape Town, South Africa) using electrochemiluminescence (ECL)-based methods. For the PK assay, a biotinylated anti-ustekinumab antibody was immobilized onto the plate, followed by the addition of the sample and a SULFO-Tag-labeled anti-ustekinumab antibody. The calibration range was 5.860–3000 ng/mL, and assay performance was confirmed with acceptable accuracy and precision across all QC levels. The ADA assay employed a bridging format. Biotinylated DMB-3115 was immobilized onto the plate, followed by sequential addition of the sample and SULFO-Tag-labeled DMB-3115. The assay demonstrated a sensitivity of 2.8 ng/mL and acceptable performance, including precision and drug tolerance. The NAb assay was conducted using a competitive ligand binding format. Biotinylated DMB-3115 was immobilized onto a streptavidin-coated plate, followed by addition of the sample and then a SULFO-Tag-labeled p40. NAb activity was assessed by the degree of signal inhibition relative to baseline. The assay demonstrated a sensitivity of 108 ng/mL and sufficient performance with respect to precision and drug tolerance.
Blood samples for immunogenicity analysis were collected on day 1 at pre-dose as well as on days 6, 14, 28, 56, and 112. An ECL assay was used for the quantification of ADAs and NAbs that reacted with DMB-3115 or ustekinumab. PK and immunogenicity analyses were conducted by Synexa Life Sciences Ltd. (Cape Town, South Africa).
Adverse events were monitored from the date when informed consent was obtained until day 112. A treatment-emergent adverse event (TEAE) was defined as an adverse event that began or worsened after administering the study drug. Laboratory tests (hematology, coagulation, clinical chemistry, urinalysis, and viral serology), vital sign measurements, and 12-lead electrocardiograms were assessed periodically throughout the study. In addition, local tolerability was assessed using the injection site reaction score (0 = none, 1 = mild, 2 = moderate, and 3 = severe) on days 1–14, 17, 21, and 28.
The primary PK endpoints were defined as area under the concentration–time curve from time zero to infinity (AUCinf), AUC from time zero to the time of the last quantifiable concentration (AUClast), and maximum serum concentration (Cmax). The secondary PK endpoints included time to Cmax (Tmax), apparent volume of distribution based on the terminal phase (Vz/F), terminal half-life (t1/2), terminal rate constant (λz), apparent total body clearance (CL/F), mean residence time from time zero to the time of the last quantifiable concentration (MRTlast), and mean residence time from time zero to infinity (MRTinf).
Statistical Analysis
The sample size was calculated under the assumption that DMB-3115 would be equivalent to reference ustekinumab (each of EU-Stelara and US-Stelara) for the primary PK endpoints (AUClast, AUCinf, and Cmax) with a true mean ratio of 1.00 between each comparison, with an interparticipant coefficient of variation (CV) of 40%. The multiplicity issue was handled by using 95% confidence interval (CI) for each comparison. In this case, 79 evaluable participants per group were needed to have the 95% CIs for the ratios of the geometric least squares means (LS means) of the primary endpoints to fall entirely within the conventional equivalence margin of 80% and 125% [13] with 90% power. Considering that participants might discontinue the study, at least 300 randomized participants (100 in each group) were required.
In the statistical analysis, the safety analysis set included all participants who received the study drug at least once, and the PK analysis set included all participants who received the study drug at least once and had evaluable primary PK endpoints without any major protocol deviation. PK parameters were calculated according to the non-compartmental methods using Phoenix® WinNonlin® version 8.2 (Certara L.P., St Louis, Missouri). In the safety analysis, adverse events were coded according to the preferred term of the Medical Dictionary for Regulatory Activities version 23.1.
In the assessment of the similarity of the PK profiles, analysis of variance was used to calculate the differences in the PK parameters between DMB-3115 and reference ustekinumab, after logarithmic transformation. In this model, study treatment was included as a fixed effect. Back transformation provided the ratio of geometric LS means (test/reference) and its 95% CI for each parameter by comparison. Equivalence for the primary PK endpoints was determined if the 95% CIs for the ratios of geometric LS means were within the equivalence margin of 80–125%.
In the post hoc analysis, we calculated the partial AUCs in the absorption and elimination phases to estimate the similarity of the PK profiles between DMB-3115 and EU-Stelara after intravenous administration. The partial AUC in the absorption phase was calculated as an AUC from time zero to the overall median Tmax (AUC0–oTmax) combined for both groups. The partial AUCs in the elimination phase were calculated as an AUC from the starting point of the elimination phase to the time of the last quantifiable concentration and an AUC extrapolated to infinity. The starting point was determined descriptively and statistically. The descriptive starting point was based on the mean concentration–time curve. A statistical starting point was the point beyond which the elimination phase was estimated to begin in 95% of participants. In the assessment of the similarity, 90% CIs for the ratios of geometric LS means of the partial AUCs were calculated, because EU-Stelara was the only comparator and multiplicity adjustment was not necessary. All data were analyzed using SAS® version 9.3 (SAS Institute, Cary, NC). The statistical differences between the treatment groups were determined with Fisher’s exact test.

Comments (0)