
Remember me
This was a randomized, double-blind, single-dose, four-group, four-period, cross-over comparative oral bioavailability study in healthy, adult human subjects under fasting conditions. We have presented the data for three groups, while the data for one group, i.e., Ashwagandha 8% (ZEN 8), is not presented in this report due to the difficulties in the preparation of technical replicates and stability associated with this product. The study was initiated after obtaining written approval from an institutional ethics committee, ‘The Research Ethics Committee’ (EC registration Number: ECR/84/Indt/TN/2013/RR-19), Coimbatore, India, on 15 October 2022. The study was carried out in compliance with the requirements of the Indian Council of Medical Research (ICMR) ethical guidelines, International Council for Harmonization (ICH) ‘Guidance on Good Clinical Practice’ (E6R2), and the Declaration of Helsinki. The study was registered with the Clinical Trials Registry of India (CTRI/2022/11/047039). Voluntary informed consent was obtained from every participant before enrolling them into the study. Subjects were randomized using SAS software (SAS® Software Version 9.4, North Carolina, USA)-generated randomization schedule to receive different products. The study used a computerized randomization process overseen by the statistician to maintain blinding. Each product bottle was labeled with a unique pre-printed code, which was the sole identifier distinguishing the formulations. Investigators dispensed the products to participants based on this randomization. Subjects were asked to consume two capsules of Ashwagandha 1.5% (ZEN 1.5) manufactured by OmniActive Health Technologies, India, with total dose of 125 mg providing 1.88 mg of total withanolides (Fig. 1) or two capsules of Ashwagandha 5% (ASH 5) with total dose of 600 mg providing 30 mg total withanolides, as declared in the certificate of analysis (COA) or two capsules of Ashwagandha 10% (ASH 10) with a total dose of 500 mg ASH 10 providing 50 mg withanolide glycosides, 160 mg of oligosaccharides, and ≤ 2.5 mg of withanolide aglycone as withaferin A, as declared in the COA. The reference products (Supplementary documents COA 2-3) used in the current study are well-established commercial products with established doses based on multiple human clinical studies. We have used the commercial product as it is available from the vendor using information from their product COA for withanolide content and published efficacious doses in our study (additional details of the study products are given in the supplement Table 2).
Fig. 1
The ZEN 1.5 used in our study is a standardized hydro-alcoholic preparation containing 1.5% total withanolides, quantified by high-performance liquid chromatography in accordance with the United States Pharmacopeia (USP) guidelines, which define seven specific peaks for total withanolide analysis [11] (see supplement Table 3). The comparator products were procured directly from a vendor along with the COAs (supplementary documents COA 1–3).
For each period, subjects were housed in the clinical facility for at least 24 h pre-dose to 24 h post-dose, and the washout period of 8 days was followed from the successive dosing day. The washout period of 8 days (equivalent to at least five elimination half-lives) was considered based on previous studies [12], indicating that the half-life for Ashwagandha is 2.27 ± 0.44 h, in order to minimize the presence of measurable drug levels before dosing in the subsequent period. In each period, after an overnight fasting of at least 10 h, two capsules of the study product were administered to the subjects as a single dose with 240 mL of drinking water as per the randomization schedule in a sitting upright posture (see supplement Fig. 1 for visual timeline of the study phases).
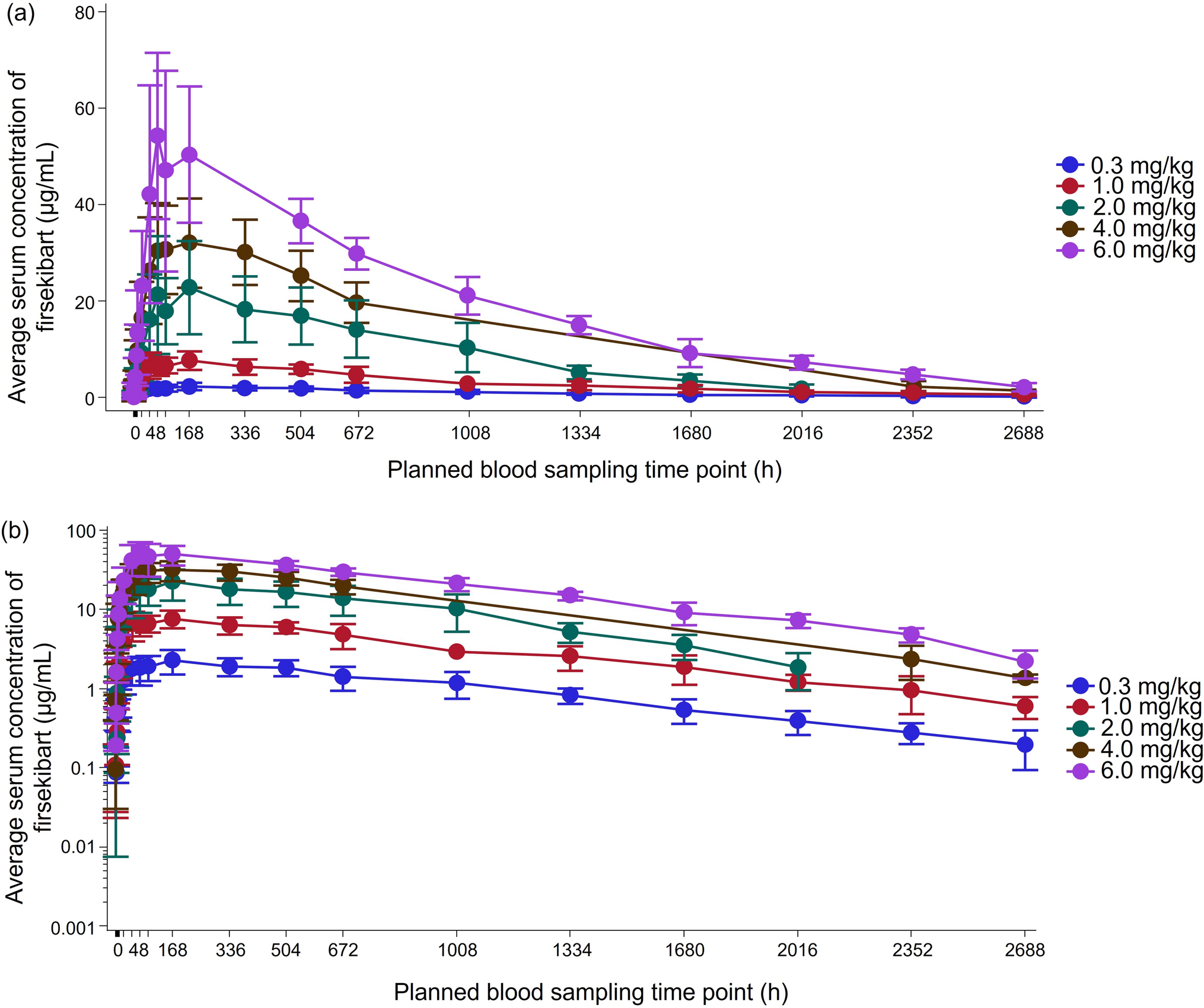
In each period, 13 blood samples (6 ml each) were collected, including the pre-dose sampling collected within 60 min prior to dosing. Post-dose blood samples were collected at 00.25, 00.50, 00.75, 01.00, 02.00, 03.00, 04.00, 05.00, 06.00, 09.00, 12.00, and 24.00 h from the dosing time. All the samples were collected in pre-labeled K2EDTA-Vacutainers from the forearm vein using an indwelling cannula.
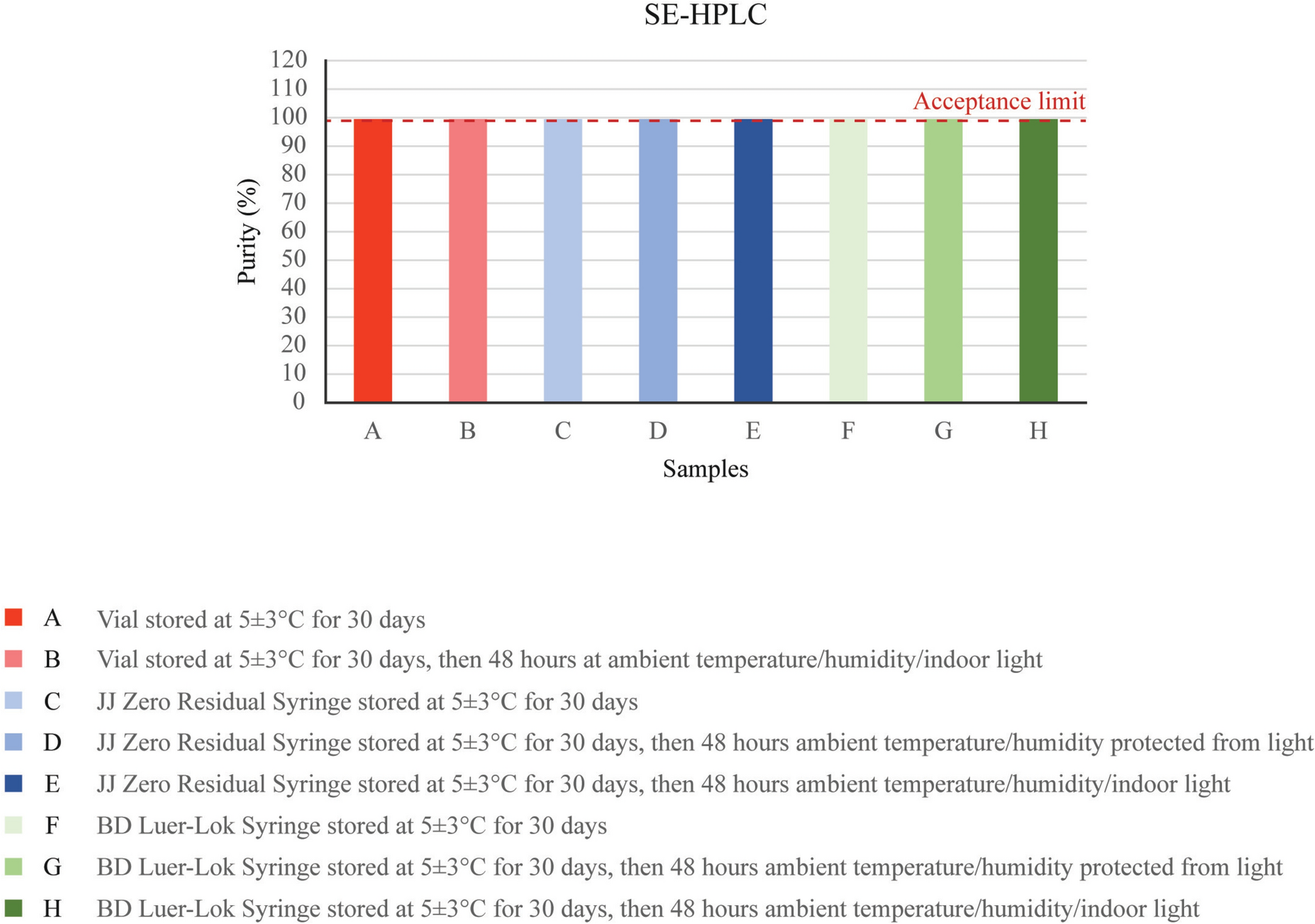
After collection of the blood samples, they were placed in a thermo-insulated box containing wet ice, and the box was transferred to the sample processing room where the blood samples were centrifuged at 4000 rpm for 10 min at 4°C to separate the plasma. Centrifugation was started within 30 min of the collection of the samples, at each collection time-point. The resulting plasma was properly labeled, stored at − 70 ± 15 °C until analysis. The concentration of total withanolides (withanoside IV, withanolide A, 12-deoxywithastramonolide, and withaferin A) in plasma was quantified by a selective and sensitive validated method using LC–MS/MS in the bioanalytical facility of Spinos Life Science and Research, Coimbatore, India (which is diagnostic laboratory services accredited by the National Accreditation Board for Testing and Calibration Laboratories, Certificate No. MC-2065, Issue date 23/04/2022, valid until 22/04/2024). Withanoside IV, withanolide A, 12-deoxywithastramonolide, and withaferin A and their internal standard, escitalopram oxalate, were selectively isolated from 500 μL of plasma by solid-phase extraction, and estimation was carried out by mass spectrometric detection on Luna® HILIC 200 Å (4.6 × 150 mm), 5-μm column. Separation was achieved by a C18 column using Buffer-2 and Acetone-M as mobile phase. The mass transition ion-pair was withanolide A: m/z 488.400 → 289.200, withanoside IV: m/z 800.500 → 459.300, 12-deoxywithastramonolide: m/z 471.300 → 299.200, and withaferin A: m/z 471.300 → 281.200 quantification, and mass transition for internal standard was 325.100 → 109.100. The method showed excellent linearity (r2 > 0.997) over the concentration ranges of 0.1560–20.8311 ng/mL, 0.1573–20.9999 ng/mL, 0.1556–20.7662 ng/mL, and 0.1540–20.5573 ng/mL for withanoside IV, withanolide A, 12-deoxywithastramonolide, and withaferin A, respectively. The software used for controlling this system and analyzing the results was Analyst software version 1.5.1 (supplied by AB SCIEX). The reference standards of the analytes and internal standard were purchased from Natural Remedies, Bangalore, India, and BioOrganics, and Applied Materials, Bangalore, India respectively.
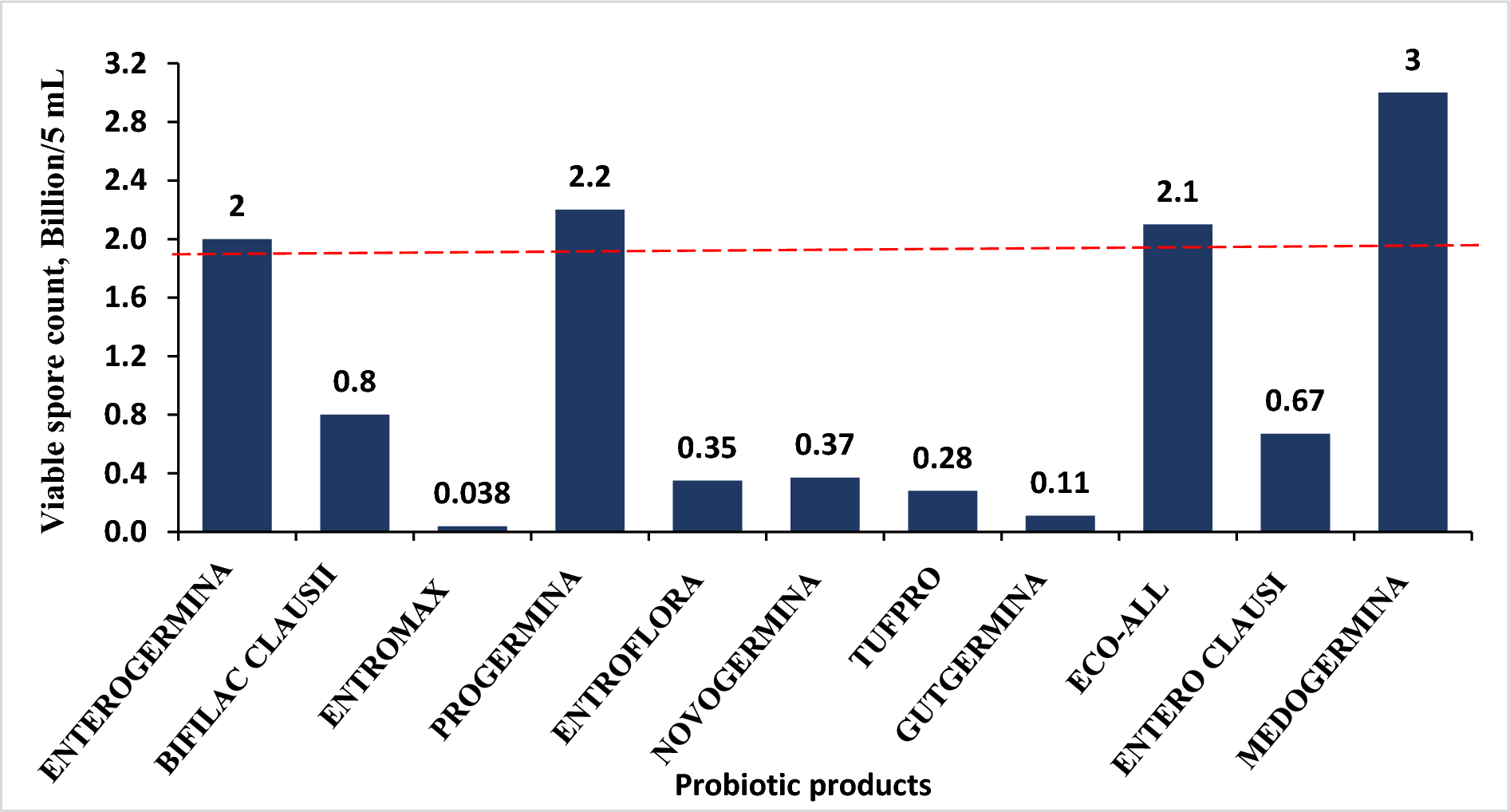
Total withanolides content was the calculated individual constituents of withanoside IV, withanoside V/VI, withaferin A, withanolide A, withanone, withanolide B, and 12-deoxywithastromonolide, as reported earlier [13]. However, our study analyzed four key withanolide compounds (withanoside IV, withaferin A, withanolide A, and 12-deoxywithastromonolide) to calculate the total withanolide content based on the availability of a validated source of reference standards at the time of analysis.
Study Population and Inclusion/Exclusion CriteriaA total of 21 healthy subjects were enrolled in the study, wherein 1 subject was kept as standby to replace any participant who might withdraw or drop out before dosing in Period I. Since all 20 enrolled subjects completed the pre-dose procedures and were successfully dosed in Period I, the standby subject was released and excluded from the study to ensure randomization integrity and maintenance of the study flow.
Volunteers who met all of the following criteria were considered for enrolment in the study: normal, healthy, adult, human subjects of age between 18–55 (both inclusive) years with a body mass index (BMI) between 20.00 and 29.99 kg/m2 (both inclusive); no evidence of underlying disease during screening and check-in and screening performed within 21 days of check-in; screening laboratory values within normal limits or considered by the physician or principal/clinical investigator to be of no clinical significance; healthy as documented by the medical history, normal physical examination, and vital signs assessments; generally healthy as documented by 12-lead electrocardiogram (ECG), chest X-ray, and clinical laboratory assessments; compliant with all requirements of this study protocol as well as instructed by the study personnel; and non-smoker.
Volunteers who met any of the following criteria were excluded from the study: evidence of allergy or known hypersensitivity to Ashwagandha or its metabolites; any significant disease or illness; history of alcohol addiction or abuse; consumed caffeine- and xanthine-containing products (i.e., coffee, tea, chocolate, and caffeine-containing sodas, colas, etc.), cigarettes, and tobacco-containing products for at least 24 h prior to check-in and throughout the entire study; consumed alcohol and its products, grapefruit and its juice, and poppy-containing foods within 48 h prior to clinic admission and throughout the entire study; consumed any prescription medications within 14 days prior to study check-in and throughout the study and any over-the-counter medicinal products, herbal medications, including Ashwagandha within 7 days prior to study check-in and throughout the study; taken an unusual diet for whatever reason (e.g., low salt) for 48 h prior to dosing and throughout the study; participated in any other study within 90 days of check-in; history of difficulty in swallowing and accessibility of veins; showed positive results for urine screen of drugs of abuse (marijuana—THC, amphetamine—AMP, barbiturates—BAR, cocaine—COC, benzodiazepines—BZD, and morphine—MOR) in urine prior to check-in of this study period, and for alcohol test (urine sample) prior to check-in of each period; underwent blood donation/excess blood loss within 90 days of check-in; ingestion of any hormonal agent at any time within 14 days prior to start of study check-in; used hormone replacement therapy for a period of 6 months prior to dosing; and females who were positive in pregnancy screening, lactating, and planning to become pregnant during conduction of the study.
Statistical AnalysisThe mean, standard deviation, minimum, median, maximum, and coefficient of variation were calculated for plasma concentrations at each individual time point as well as for the pharmacokinetic parameters (Cmax, AUC0-t, Tmax, and t½) for baseline-corrected total withanolides (sum of withanoside IV, withanolide A, 12-deoxywithastramonolide, and withaferin A). In addition, geometric LSM, ratio of means, and 90% confidence intervals were provided for (Cmax, AUC0-t, Tmax, and t½) baseline-corrected total withanolides determined by using Phoenix WinNonlin Version 8.3.3. Statistical comparisons were made between ZEN 1.5 and ASH 5, and ZEN 1.5 and ASH 10 on the individual time points and pharmacokinetic parameters by ANOVA using SAS® software Version 9.4 (SAS Institute, NC, USA). A P value of < 0.05 was considered as statistically significant.
Sample Size DeterminationConsidering a significance level of 5%, a power of 80%, and a sample size of 16 subjects was sufficient to establish bioavailability comparisons between the four Ashwagandha formulations under fasting conditions with an adequate power. However, considering the dropout or withdrawals due to personal reasons, 20 subjects were randomized and dosed.
Safety AnalysisSafety assessments included monitoring of adverse events, physical examination, vital signs measurements, and laboratory assessments. An ECG was recorded at screening and 24 h before check-out from the study. Monitoring for adverse events was carried out throughout the study period.
Comments (0)