
Remember me
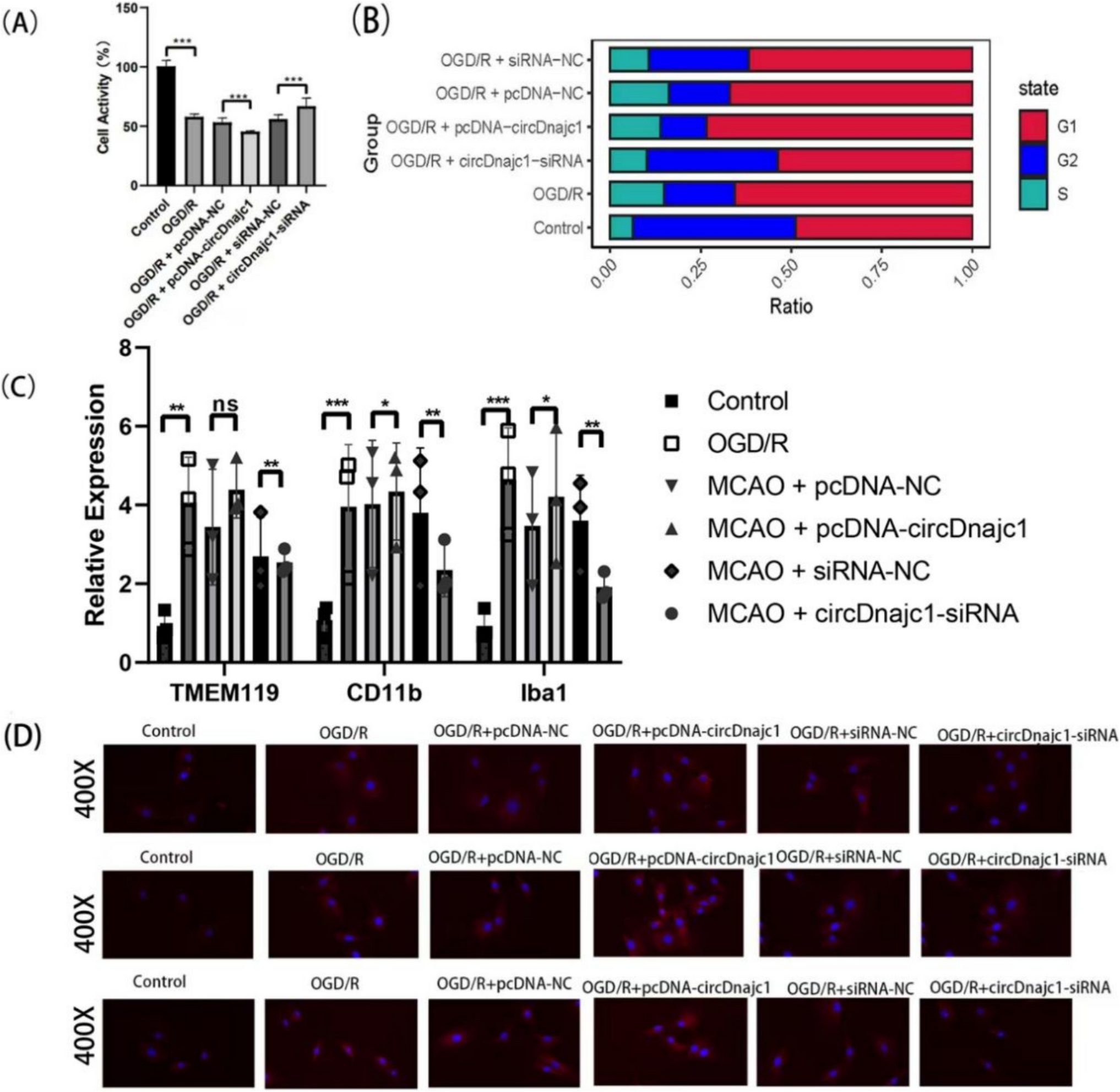


Metformin is an orally active drug, though it has low lipid solubility and is less absorbed from the intestines [13,14,15]. Thus, the bioavailability of metformin is about 50% of the administered dose [16]. Metformin requires specific organic cationic transporters (OCT) for its absorption from the intestines. Metformin absorption via OCT is a saturable process; therefore, there is an inverse association between the administered doses and the intestinal absorption rate [17,18,19,20]. Metformin is not metabolized and is mainly excreted by the kidney [21]. In addition, metformin has minimal plasma protein binding affinity. Thus, it has a short half-life of about 4–5 h [22]. Metformin is regarded as a safe drug and is less susceptible to drug-drug interaction [22]. However, prolonged and toxic doses of metformin may lead to the development of lactic acidosis, especially in patients with heart failure and renal impairment. Metformin can cross the blood–brain barrier (BBB) but needs time to produce the anorexigenic effect in diabetic patients [23].
The fundamental mechanism of metformin’s action is not fully clarified. However, the hypoglycemic effect of metformin is related to inhibiting gluconeogenesis, reducing intestinal absorption of glucose, and increasing peripheral insulin sensitivity [24]. The cellular effect of metformin is mediated by activating the expression and signaling pathways of AMPK, which is required for the inhibitory effect of metformin on hepatocyte production of glucose [24]. However, metformin can produce many pleiotropic and cellular impacts independent of the AMPK signaling pathway [25]. Metformin inhibits mitochondrial complex I, interfering with the mitochondrial respiratory chain, reducing cellular ATP, and increasing AMP with subsequent activation of AMPK [25].
Interestingly, AMPK activation reduces gluconeogenesis when the energy substrates are reduced [26, 27]. However, the neuronal store of nutrients is inattentive due to the absence of a glycolytic pathway. Therefore, a stressful condition such as ischemic stroke induces the over-activation of the AMPK signaling in the brain following ischemic stroke [28]. In addition, metformin inhibits mitochondrial glycerophosphate dehydrogenase, inhibiting hepatic gluconeogenesis [27]. Furthermore, metformin can bind copper ions, an essential metal for activating mitochondrial enzymes and ATP production [29]. Thus, the metal-binding effect of metformin could be an alternative mechanism for the action of metformin [30].
Furthermore, metformin inhibits different enzymes, such as fructose 1,6-bisphosphatase 1 (FBP1) and adenylate cyclase, which are involved in gluconeogenesis by increasing AMP in the hepatocytes [27]. By increasing the AMP/ATP ratio, metformin promotes the activation of AMPK [27]. Metformin also inhibits mitochondrial glycerol phosphate dehydrogenase (mGPDH), increasing the NADH:NAD ratio and reducing the gluconeogenesis process from lactate [31]. Also, metformin increases the hepatic redox state by regulating the expression of glutathione (GSH) and oxidized glutathione (GSSG), resulting in the inhibition of many genes involved in the gluconeogenesis process [32]. Metformin, by inhibiting mitochondrial respiratory chain complex IV, indirectly suppresses the activity of mGPDH. Metformin can affect the lysosomal action by inhibiting v-ATPase independent of the AMPK pathway [33]. Besides, metformin activates liver kinase B1 (LKB1), induces the release of glucagon-like peptide 1 (GLP1) from the intestines, and regulates different genes involved in glucose homeostasis [34]. Therefore, the mechanism of metformin’s action is complex and related to other signaling pathways (Fig. 1).
Fig. 1
The mechanism of metformin: Metformin activates AMP-activated protein kinase (AMPK) in the liver, which leads to suppression of fatty acid synthesis and gluconeogenesis. Metformin also activates AMPK in skeletal muscle, increasing the translocation of glucose transporter 4 to the cell membrane and thereby increasing glucose uptake. Increased AMPK also inhibits mitochondrial glycerol-3-phosphate dehydrogenase (mGPD), leading to an increase in cytosolic NADH, which both stimulates the conversion of pyruvate to lactate and decreases gluconeogenesis. SREBP-1c, sterol regulatory element binding protein-1c; PEPCK, phosphoenolpyruvate carboxykinase; GAPase, glucose 6-phosphatase; GLUT 4, glucose transporter 4
Moreover, metformin reduces cellular energy by inhibiting mitochondrial oxidative phosphorylation with subsequent inhibition of gluconeogenesis, depending on the cellular ATP [35]. Metformin modulates gut microbiota and attenuates T2D-induced gut dysbiosis [36]. The regulation of gut microbiota may mediate the glucose-lowering effect of metformin [37]. It has been shown that depletion of gut microbiota attenuates the glucose-lowering effect of metformin [38]. Supporting this finding, restoration of gut microbiota by microbiota transplant improves the glucose-lowering effect of metformin [39, 40]. Metformin-induced alteration of gut microbiota induces the expression of different genes involved in regulating blood glucose, such as the GLP1 gene [40]. It has been shown that metformin activates the release of the short-chain fatty acids (SCFAs) from gut microbiota that induce the expression of the GLP1 gene in the enteroendocrine L cells with subsequent release of GLP1 hormone [41]. Modulation of gut microbiota by metformin enhances the synthesis and release of bile acids, improving glucose homeostasis by inhibiting intestinal farnesoid X receptor (FXR) through an AMPK-independent mechanism [42]. Moreover, metformin improves the expression of the GDF15 gene, enhancing insulin sensitivity and having anti-inflammatory effects [43]. The central effect of metformin, mainly the anorexigenic effect, is mediated by the activation of GDF15 [44]. Therefore, through activation of AMPK, GDF15, and other signaling pathways, metformin may affect the neuropathology of ischemic stroke.
Ischemic Stroke: An OverviewIschemic stroke is defined as a focal neurological deficit caused by embolism or thrombosis of cerebral vessels of particular brain areas [45]. Ischemic stroke represents about 87% of total stroke type [46]. Ischemic stroke is the foremost cause of long-lasting disability and death internationally [47]. Diverse risk factors are concerned with the progress of ischemic stroke, including non-modifiable and modifiable risk factors [48,49,50,51,52,53]. Initial diagnosis of ischemic stroke is crucial as the thrombolytic treatments lead to noteworthy enhancement when administered earlier, after the onset of ischemic stroke [54]. Rising evidence from different studies demonstrated that ischemic stroke is linked with the development of oxidative stress and neuroinflammation due to unrestrained activation of inflammatory signaling pathways and the generation of reactive oxygen species (ROS) [55]. These variations may influence the development of neurodegeneration and the progression of post-stroke seizure and neurodegenerative diseases [56].
Ischemic stroke and related brain hypoxia disturb mitochondrial function, tricarboxylic acid cycle, and glycolysis, with subsequent disruption of the neuronal bioenergetics process [57]. It has been revealed that ATP exhaustion after ischemic stroke promotes the molecular and deleterious cellular events recognized as the ischemic cascade [58]. Furthermore, the absence of ATP within a few minutes before ischemic stroke activates irregular ionic efflux across the neuronal membrane [58]. It has been shown that low ATP deregulation of neuronal membrane Na +/K + ATPase decreases glutamate uptake [59]. In addition, impairment of glutamate clearance through glutamate transporters located in the neurons and glial cells is developed due to the inhibition of Na+/K+ ATPase, resulting in glutamate accumulation and neuronal excitotoxicity development [59].
Furthermore, overactivation of postsynaptic glutamate receptors by glutamate in the synaptic cleft activates calcium overload by stimulating voltage-gated calcium channels. It activates many enzymes, such as nuclease and protease [60]. Besides, cytoplasmic calcium overload through induction of mitochondrial dysfunction augments the generation of oxidant molecules, which prompt the release of nitric oxide and peroxidation of neuronal cardiolipin [61].
Similarly, a decrease in neuronal ATP reduces the production of intracellular antioxidant enzymes, with subsequent exaggeration of neuronal injury induced by ischemic-hypoxic changes in ischemic stroke [62]. Also, brain ischemia and ischemic reperfusion injury trigger the activation of neuronal phospholipase A2 (PLA2), which prompts hydrolysis of membrane phospholipid and the release of free fatty acids and lysophospholipids, which induce the development of the pro-inflammatory process in ischemic stroke [63]. Henceforth, the pathogenesis of ischemic stroke is complex and connected to the release of pro-inflammatory cytokines and the progression of oxidative stress (Fig. 2).
Fig. 2
The pathogenesis of ischemic stroke: Ischemic stroke and brain hypoxia interrupt the neuronal bioenergetics process. Absence of ATP in ischemic stroke activates irregular ionic efflux across the neuronal membrane, causing abnormal neuronal membrane Na +/K + ATPase, decreases glutamate uptake, impairment of glutamate clearance resulting in the accumulation of glutamate, and the development of excitotoxicity. Overacting postsynaptic glutamate receptors by stimulating voltage-gated calcium channels initiates many enzymes, such as nuclease and protease enzymes. Cytoplasmic calcium overload through the initiation of mitochondrial dysfunction augments the generation of oxidant molecules, which prompt the release of nitric oxide and peroxidation of neuronal cardiolipin. A decrease in neuronal ATP lessens the production of intracellular antioxidant enzymes, with subsequent exaggeration of neuronal injury induced by ischemic-hypoxic changes in ischemic stroke. Brain ischemia activates neuronal PLA2 (phospholipase A2), which prompts hydrolysis of membrane phospholipid and the release of free fatty acids and lysophospholipids, which induce the development of the pro-inflammatory process in ischemic stroke
The Role of Metformin in Ischemic StrokeIt has been suggested that metformin could be effective against the development and progression of ischemic stroke and neurodegenerative diseases by regulating the functional activity of the brain neurovascular unit (NVU) [8, 64,65,66]. Metformin affects various NVU components, including pericytes, astrocytes, microglia, and vascular endothelial cells, mainly serving to protect the BBB [66]. Regulating the inflammatory response in NVU may be the primary mechanism of metformin in improving CNS-related diseases. Thus, metformin may be a potential drug for treating diseases associated with NVU deterioration. Mounting evidence from preclinical studies highlighted the protective role of metformin against the development of ischemic stroke. The administration of 50 mg/kg/day of metformin for 24 h following middle cerebral artery occlusion for 3 weeks improved neurogenesis and reduced ischemic stroke severity in mice [67]. However, in AMPK α−2 knockout mice, metformin treatment has no beneficial effects on recovery and angiogenesis, suggesting that metformin-induced angiogenic effects are mediated by AMPK [67]. Studies indicated that increasing cortical neurogenesis correlates with functional recovery following ischemic stroke [68, 69]. Angiogenesis stimulation generates new vessels to improve collateral circulation. Intrinsic genetic mechanisms and growth factors control neurogenesis. The leading process of the migrating neural progenitor cells (NPCs) is closely associated with blood vessels, suggesting that this interaction provides directional guidance to the NPCs. These findings suggest that blood vessels play an important role as a scaffold for NPC migration toward the damaged brain region [68, 69]. It has been demonstrated that angiogenesis only occurred transiently in the cortex of the ischemic hemisphere, implying that the new vessels were merely part of the clean-up after stroke rather than a contribution to neurorestoration [68, 69]. Also, induction of neurogenesis in the ischemic stroke penumbra region plays a role in eliminating cellular debris after ischemic stroke [70]. Metformin improves neuronal repair after ischemic stroke by increasing the differentiation of neural precursor cells into neuronal cells in mouse ischemic models [71, 72]. Metformin treatment also meaningfully reduced the infarct volume and alleviated functional dysfunction after stroke. Mechanistically, metformin promoted NPC migration via upregulating the CDC42 gene expression [68]. Therefore, metformin represents an optimal candidate agent for neural repair capable of expanding the adult NPC population and subsequently driving them toward the destination for neuronal differentiation.
Moreover, metformin reduces the severity of cerebral ischemic reperfusion injury by inhibiting mitochondrial dysfunction, oxidative stress, neuroinflammation, impairment of BBB, and neuronal apoptosis [14, 65, 72,73,74,75,76]. Zeng et al. [77] found that metformin protects against cerebral ischemic-reperfusion injury by regulating the expression of long non-coding RNA and Rho-associated protein kinase 2 (ROCK 2), which has a neurodetrimental impact on the pathogenesis of ischemic stroke [78, 79]. Metformin can significantly alleviate acute and chronic cerebral ischemic reperfusion injury, and it has a strong regulatory effect on stroke-induced oxidative stress. The expression of lncRNA-H19 and ROCK2 could be downregulated with metformin in vivo and in vitro [77]. Thus, metformin exerts neuroprotective effects by regulating ischemic stroke-induced oxidative stress injury via the lncRNA-H19/miR-148a-3p/ROCK2 axis. These results provide new evidence that metformin may represent a potential treatment for stroke-related brain injury. In ischemic stroke, the AMPK signaling is downregulated, leading to microglia polarization and neuronal loss [11, 80]. Thus, through activating AMPK signaling, metformin reduces ischemic stroke severity in the experimental stroke models [81, 82]. It has been shown that metformin upregulated the brain-derived neurotrophic factor (BDNF) expression and increased phosphorylation levels of AMPK and cAMP-response element binding protein (CREB) in the ischemia penumbra. This effect of metformin is reversed by Compound C [81, 82]. Therefore, metformin improves BDNF expression in the cerebral ischemic penumbra via the activation of the AMPK/CREB pathway, thereby protecting cerebral ischemic reperfusion injury.
Furthermore, metformin at 20 mg/kg improved neurological function and attenuated brain edema, oxidative stress, and BBB permeability disruption 24 h after subarachnoid hemorrhage, promoted mitophagy in an AMPK-dependent manner. Metformin attenuated early brain injury after subarachnoid hemorrhage in rats through AMPK-dependent signaling [81, 82]. These protective effects might be achieved by regulating mitochondrial morphology and promoting mitophagy [64, 65]. Furthermore, due to its anti-inflammatory and antioxidant properties, metformin attenuates the pathogenesis of ischemic stroke in animal models [81, 83]. Metformin, by promoting the expression of neuroprotective BDNF and antioxidant genes, inhibits the expression of inflammatory signaling pathways [81, 83]. Moreover, metformin encourages the activation of AKT1 and reduces the phosphorylation of JNK3 and c-Jun and elevation of cleaved caspase-3 in ischemia/reperfusion brains [14, 74]. PI3 K inhibitor reversed all the protective effects [81, 82], indicating that metformin protects the hippocampus from ischemic damage through PI3 K/Akt1/JNK3/c-Jun signaling pathway [84]. A meta-analysis of preclinical studies highlighted that administration of metformin attenuates the pathogenesis of ischemic stroke and improves the functional recovery of neurological deficits after ischemic stroke [84]. Notably, metformin treatment plays a neuroprotective role and improves pathological and behavioral outcomes in rodent stroke models [76, 85]. Although multiple
Comments (0)