
Remember me



To delineate the cellular heterogeneity of LUAD, we analyzed the scRNA-seq data from 21 patients, including 18 LUAD tumor tissues and 18 adjacent normal tissues. After quality control, dimensionality reduction, and batch effect correction, a total of 10 major cell populations were identified and visualized using UMAP (Fig. 1A). These populations were annotated as epithelial cells, fibroblasts, myeloid cells, T cells, B cells, NK cells, plasma cells, endothelial cells, mast cells, and neutrophils based on canonical marker gene expression.
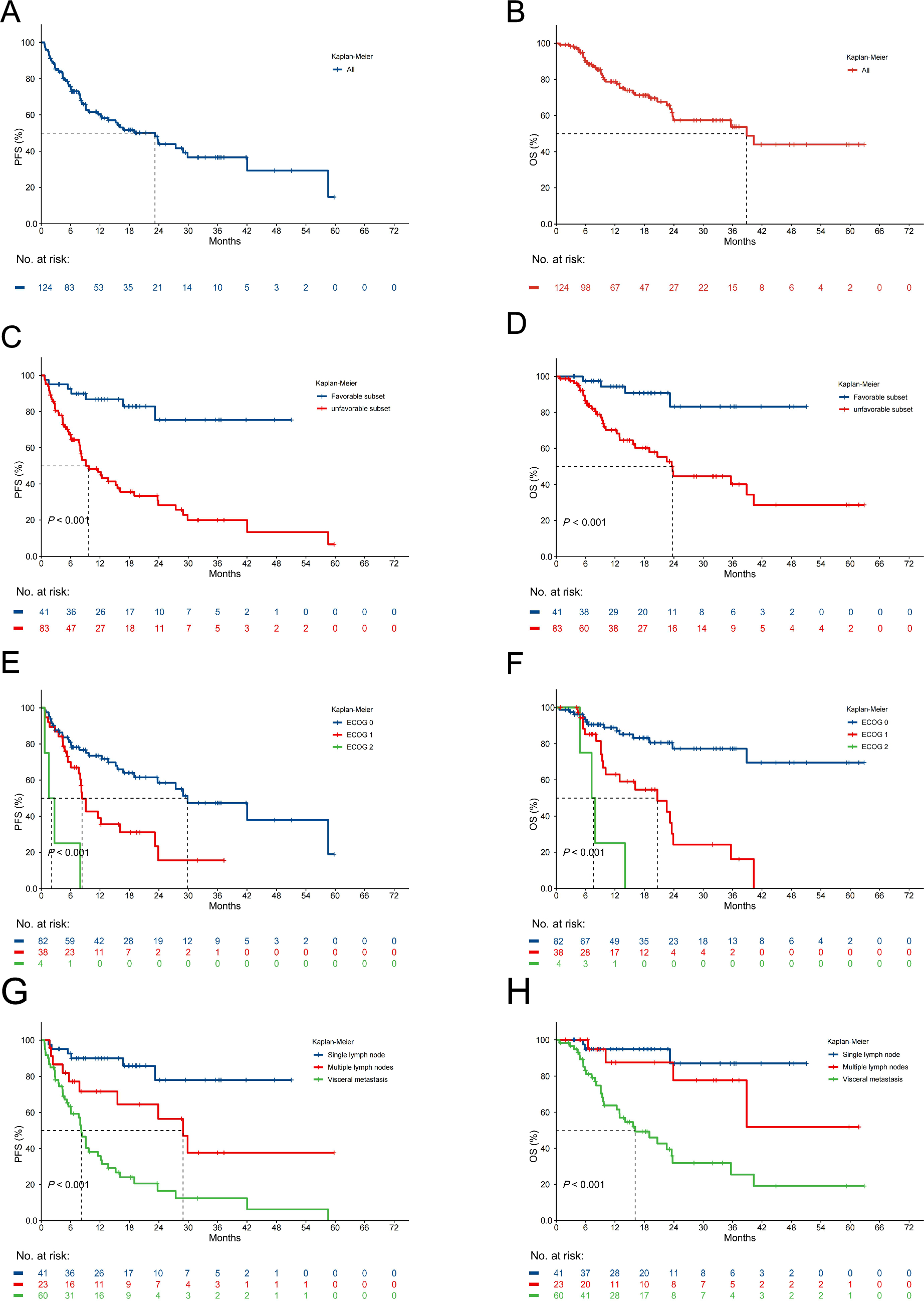
Fig. 1
Single-cell atlas of lung adenocarcinoma and adjacent normal tissues. A Uniform manifold approximation and projection (UMAP) visualization of 10 major cell populations in LUAD. B Stacked bar plot showing the distribution of 10 cell types across 21 patients. C Bar plots comparing cell type proportions between tumor (left) and normal tissues (right). D Bubble plot displaying marker genes for each cell type. E Feature plots illustrating spatial distribution of marker genes in UMAP space
The distribution of these 10 cell types across individual patients was consistent, although certain cell types displayed inter-patient variability (Fig. 1B). We next compared the cellular composition between tumor and normal tissues. As shown in Fig. 1C, tumor tissues exhibited a marked enrichment of epithelial cells, T cells and myeloid cells, whereas immune populations (such as T cell and B cells), and myeloid cells were relatively more abundant in normal tissues.
To further validate the identified cell types, we analyzed the expression patterns of known marker genes. A bubble plot summarized the top markers for each cell population, confirming their identity (Fig. 1D). Furthermore, feature plots illustrated the spatial distribution of key marker genes within the UMAP space, highlighting distinct transcriptional profiles across different cell types (Fig. 1E). These findings collectively provide a comprehensive single-cell atlas of LUAD tissues and their adjacent normal counterparts.
Heterogeneity and developmental potential of epithelial cellWe next performed the deep clustering of epithelial cells, revealing eight distinct subpopulations (MUC21 + Epi, SFTPC + Epi, MKI67 + Epi, C11orf97 + Epi, TMEM45A + Epi, RTKN2 + Epi, ASCL1 + Epi, and TFF2 + Epi) through UMAP visualization (Fig. 2A). Marker gene analysis confirmed distinct epithelial subpopulations: SFTPC+ (alveolar-like), MKI67+ (proliferative), ASCL1+ (neuroendocrine-like), and tumor-enriched TMEM45A+ (extracellular matrix remodeling) and RTKN2+ (cell adhesion), underscoring functional specialization within the tumor microenvironment (Fig. 2B).
Fig. 2
Epithelial cell heterogeneity and differentiation dynamics. A UMAP plot of eight epithelial subclusters. B Bubble plot showing marker genes for epithelial subpopulations. C Bar plots comparing epithelial subset proportions (left) and cell counts (right) between tumor and normal tissues. D Violin plots of copy number variation (CNV) scores across epithelial subclusters. E Ro/e index heatmap depicting tissue-specific enrichment (red: tumor; blue: normal). F–G Potency score distribution and bar plot highlighting differentiation potential. H Trajectory analysis illustrating five evolutionary paths of epithelial subsets
Tumor tissues showed a pronounced expansion of MUC21 + Epi and MIK67 + Epi subpopulations, while SFTPC + Epi and C11orf97 + Epi subsets were relatively enriched in normal tissues (Fig. 2C). CNV scores further distinguished malignant subpopulations, with MUC21 + Epi and ASCL1 + Epi cells displaying elevated CNV levels, indicative of genomic instability (Fig. 2D). Ro/e index analysis highlighted the tumor-specific enrichment of TFF2 + Epi and MUC21 + Epi subpopulations, contrasting with the normal tissue dominance of RTKN2 + Epi and SFTPC + Epi subsets (Fig. 2E).
Developmental potential analysis revealed heterogeneous potency scores across subpopulations, with SFTPC + Epi exhibiting unipotent-like states, while C11orf97 + Epi subsets were predominantly differentiated (Fig. 2F–H). Trajectory inference delineated five evolutionary paths, linking SFTPC + Epi subpopulations to various differentiated states (RTKN2 + Epi, MKI67 + Epi and C11orf97 + Epi), suggesting dynamic plasticity during tumor progression (Fig. 2I). Collectively, these findings uncover the molecular and functional diversity of epithelial subpopulations, implicating specific subsets in LUAD pathogenesis.
Heterogeneity and prognostic significance of fibroblast subpopulationsClustering of fibroblasts identified nine distinct subsets (Fb_MFAP5, Fb_GPC3, Fb_IGFBP4, Fb_CD52, Fb_RERGL, Fb_COL11A1, Fb_HIGD1B, Fb_WIF1, Fb_SERPINB2) with unique spatial distributions in UMAP (Fig. 3A). These subsets exhibited marker gene expression profiles (Fig. 3B), including MFAP5 (matrix organization), GPC3 (Wnt signaling), and IGFBP4 (metabolic regulation). Tumor tissues exhibited a significant increase in Fb_COL11A1 and Fb_IGFBP4 subpopulations, while Fb_MFAP5 and Fb_GPC3 were enriched in normal tissues (Fig. 3C).
Fig. 3
Fibroblast subpopulations and prognostic significance. A UMAP plot of nine fibroblast subsets. B Bubble plot of fibroblast marker genes. C Bar plots comparing fibroblast subset proportions (left) and cell counts (right). D–F Enrichment analysis for Fb_MFAP5, Fb_GPC3, and Fb_IGFBP4. G Kaplan-Meier survival curves in lung adenocarcinoma patients with high and low Fb_IGFBP4 group
Functional enrichment analysis revealed subtype-specific roles: Fb_MFAP5 was linked to extracellular matrix organization and extracellular matrix organization (Fig. 3D), Fb_GPC3 to epithelial cell migration and cell − cell signaling by wnt (Fig. 3E), as well as Fb_IGFBP4 to extracellular structure organization and Golgi transport (Fig. 3F). Strikingly, LUAD patients with high Fb_IGFBP4 group showed markedly worse survival compared to the low group in LUAD (p = 0.0024), implicating this subset in tumor progression (Fig. 3G). These findings delineate fibroblast heterogeneity and highlight prognostically relevant subpopulations in LUAD.
Tumor-specific MUC21 + Epi-fibroblast crosstalkIntercellular communication analysis revealed significant differences in interaction numbers between tumor and normal tissues, with tumor samples showing heightened CD99, FN1, COLLGENA and so on (Fig. 4A and S1A). Ligand-receptor pair analysis identified tumor-specific enrichment of fibroblast-epithelial crosstalk, notably between Fb_COL11A1/Fb_IGFBP4 and MUC21 + Epi subset, driven by different interactions, such as COL1A1/SDC4 and COL1A2/SDC4 (Figs. 4B and S1B).
Fig. 4
Tumor-specific epithelial-fibroblast crosstalk. A Bar plots comparing interaction numbers between tumor (top) and normal tissues (bottom). B Bubble plots showing differential ligand-receptor interactions between fibroblast subsets and MUC21 + Epi. C Circos plots depicting MK pathway interactions from fibroblast subsets (senders) to MUC21 + Epi (receivers) in tumor (top) versus normal tissues (bottom). Line thickness reflects interaction strength. D Circos plots illustrating COLLAGEN pathway interactions between epithelial subclusters and fibroblast subsets in tumor (top) versus normal tissues (bottom)
Pathway-centric mapping highlighted tumor-dominant MK signaling from fibroblasts (Fb_COL11A1, Fb_IGFBP4, Fb_SERPINB2) to MUC21 + Epi subset, with thickened interaction lines indicating enhanced pro-tumorigenic signaling (Fig. 4C). In addition, COLLAGEN pathway interactions between epithelial cells and fibroblasts (Fb_COL11A1, Fb_IGFBP4) were amplified in tumors, implicating matrix remodeling in invasion (Fig. 4D). Reverse MK signaling from MUC21 + Epi to fibroblasts further underscored the dynamic reciprocity of tumor-stroma crosstalk (Fig. S1C). These findings delineate a rewired interaction landscape in tumors, driven by fibroblast-epithelial synergy and pathway-specific activation.
Metabolite-mediated fibroblast-epithelial crosstalk and spatial architectureMetabolite-mediated crosstalk between fibroblast and epithelial subpopulations was markedly amplified in tumors. Comparative analysis revealed a significant increase in metabolite-driven interactions involving ASCL1 + Epi, MKI67 + Epi, TFF2 + Epi, and Fb_COL11A1 in tumor tissues, whereas normal tissues showed the main metabolite-driven interactions in MKI67 + Epi, C11orf97 + Epi, ASCL1 + Epi, and Fb_MFAP5 (Fig. 5A). Network visualization further demonstrated stronger and more complicated interaction intensities in tumors (Fig. S2A). These findings suggest a tumor-specific reliance on metabolic crosstalk to fuel epithelial-stromal crosstalk.
Fig. 5
Metabolic crosstalk and spatial architecture. A Bar plots comparing metabolite-mediated interactions between tumor (left) and normal (right) tissues. B–D UMAP clustering (B), spatial mapping (C), and co-localization (D) in LUAD tissues. E–G Normal tissue counterparts (E UMAP; F spatial mapping; G co-localization)
Deconvolution mapping highlighted heterogeneous cellular distributions in tumors, with green spots representing raw spatial transcriptomic data and red overlays aligning single-cell reference profiles (Fig. S2B). UMAP visualization and co-localization analysis of spatial transcriptomes further identified dense interactions between epithelial cell subsets (mainly MKI67 + Epi and MUC21 + Epi) and fibroblasts in LUAD tumor tissues (Fig. 5B–D). In contrast, normal lung tissues exhibited uniform UMAP clustering (Fig. 5E), homogeneous spatial mappings (Fig. 5F), and minimal stromal-epithelial overlap (Fig. 5G), underscoring tumor-specific rewiring of cellular ecosystems.
Prognostic value of MCI score and molecular signatures in LUADTo confirm that the three key subgroups of MUC21 + Epi, Fb_COL11A1, and Fb_IGFBP4 are not only associated with LUAD, but also closely related to the survival of patients with other types of NSCLC as well, we collected data from TCGA LUAD and LUSC patients to construct prognostic models. Prioritizing highly expressed genes from three critical subpopulations (MUC21 + Epi, Fb_COL11A1, and Fb_IGFBP4), we identified six survival-associated candidates. Multi-variable Cox regression found six survival-associated genes (ADAM10, MARVELD1, IER5L, MYLIP, PELI1, ANKRD65), with ADAM10 (HR = 1.716, p = 0.0044) and MARVELD1 (HR = 1.348, p = 0.0415) showing the strongest prognostic relevance (Fig. 6A). The MCI score, constructed from the expression profiles of these six genes, was statistically correlated with T stage, N stage and clinical stage, among which higher MCI score predicted aggressive clinicopathological features in general (Fig. 6B).
Fig. 6
Prognostic value of the MCI score. A Forest plot of six survival-associated genes derived from critical epithelial and fibroblast subpopulations using multivariable Cox regression. B Violin plots linking MCI score to TNM stage and clinical stage. C–D Univariate/multivariate Cox analysis of prognostic factors. E Kaplan-Meier curves stratified by MCI score (high vs. low, p < 0.0001) in lung adenocarcinoma/lung squamous cell carcinoma. F Nomogram integrating MCI score and clinical variables for survival prediction. G Time-dependent ROC curves (1-/3-/5-years)
Univariate and multivariate analyses confirmed MCI score as an independent prognostic factor (HR > 1, p < 0.001), outperforming traditional variables like TNM stage (Fig. 6C–D). To verify whether MCI scores accurately stratify patients, we performed a survival analysis, using 0.95 as a threshold to divide patients into groups with high MCI scores and low MCI scores. Patients with high MCI score exhibited markedly worse overall survival compared to the low-score group in LUAD and LUSC (p < 0.0001; Fig. 6E). A nomogram integrating MCI score, TNM stage, gender, and age provided robust 1-/3-/5-year survival predictions (Fig. 6F), with time-dependent AUC values all more than 0.6, further validating its clinical utility (Fig. 6G). These results position MCI score as a potent biomarker for NSCLC risk stratification and prognosis.
Comments (0)