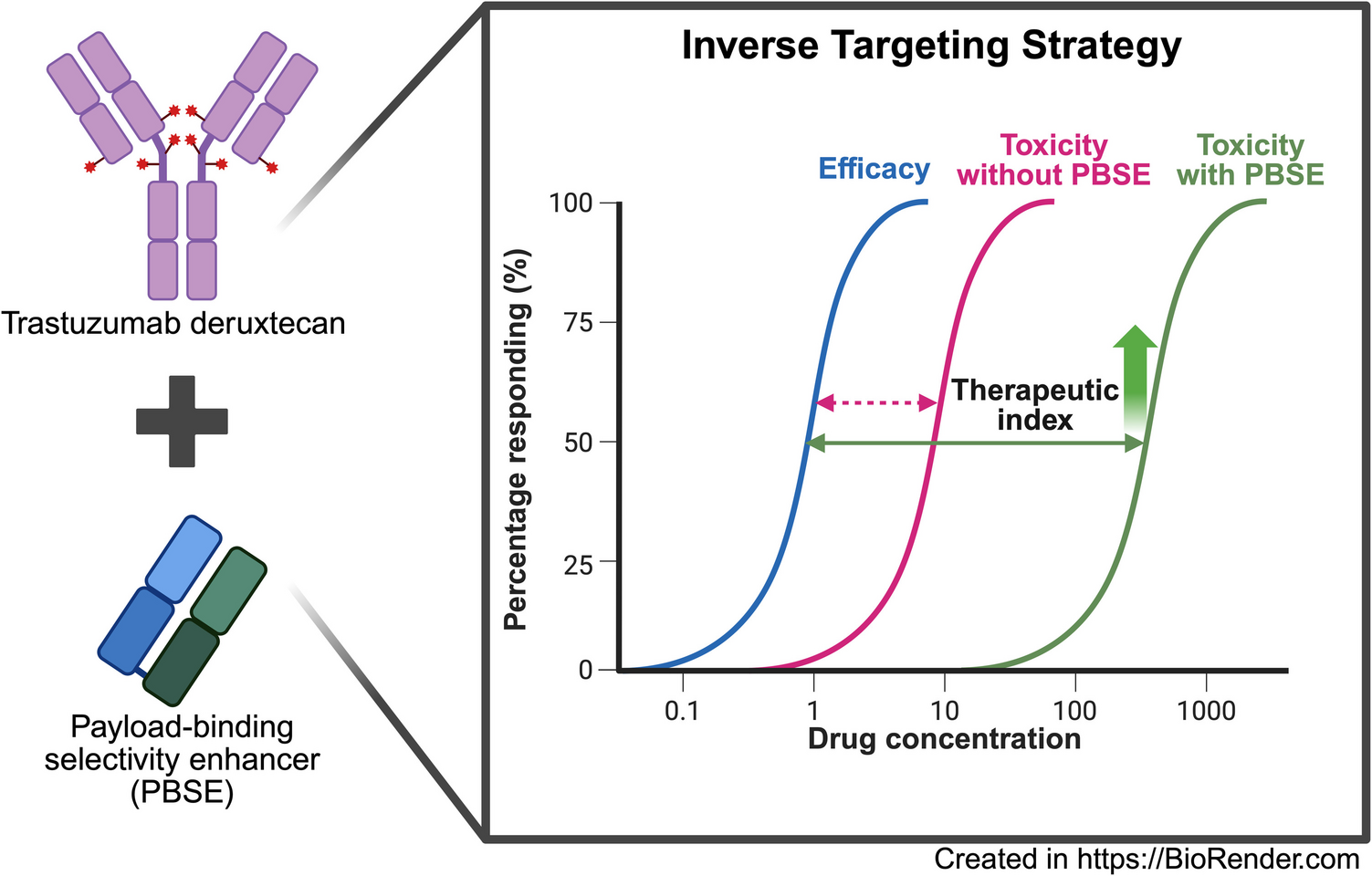
Antibody drug conjugates are designed to improve the therapeutic index of small molecule cytotoxic agents by targeting delivery of payload to cells that overexpress tumor-associated antigens. Despite the clinically proven success of several ADCs, toxicity limits tolerable ADC doses to levels below those needed to eradicate cancer in most patients. The clinical management of ADC treatment-induced toxicity include vigilant monitoring of patients, supportive care, and reduction in dose or dosing frequency (26, 27).
DXd and SN38 are CPT that inhibit topoisomerase I with intermediate cell killing potency. Their conjugation to antibodies targeting HER-2 and Trop-2 led to the regulatory approval of T-DXd and sacituzumab govitecan. The most common clinical toxicities of CPT ADCs are neutropenia, anemia, and diarrhea, which are also observed in patients treated with small-molecule CPTs such as irinotecan, exatecan, and topotecan (28, 29). The Food and Drug Administration (FDA) approved dosage of T-DXd, 5.4 mg/kg every 21 days, is based on the balance of safety and efficacy (30). During phase 1 dose expansion investigations, a positive efficacy exposure–response (E-R) relationship was observed for T-DXd at 5.4, 6.4, and 7.4 mg/kg, where higher dose was associated with longer progression-free survival. However, safety E-R analyses also showed a positive relationship between exposure and key adverse events including interstitial lung disease (ILD) (9). In pursuit of increasing the therapeutic index of ADCs, much effort has been placed on the optimization of antibody affinity and structure, selection of different types of anticancer payloads, and advancements in linker chemistry, leading to several generations of ADCs (14, 31, 32). However, optimization of linker chemistry, antibody-to-drug ratio, and payload potency of each individual ADC under development is a complex and resource-intensive challenge.
Co-administration of payload-binding selectivity enhancers (PBSE) with ADCs does not necessitate any modification or alteration to the structure of ADCs and, consequently, the inverse targeting strategy that we have proposed may allow rapid enhancement of the therapeutic index of many ADCs, including those already approved and those under clinical development. The administration of binding agents to mitigate or reverse the unwanted effects caused by drugs such as digoxin and dabigatran or toxin has been well-established clinically (33,34,35). Additionally, our lab has demonstrated the application of anti-drug antibodies in inverse targeting strategies to alter the pharmacokinetics of several chemotherapeutics including methotrexate and topotecan, resulting in decreased drug-related toxicity and improved therapeutic selectivity (20, 22, 36). Our recent work demonstrates that the inverse targeting strategy may be applied to optimize ADC safety and efficacy. For example, we have demonstrated co-administration of anti-MMAE PBSE decreased myelosuppression and weight loss following dosing of MMAE-based ADCs while not negatively affecting anti-tumor efficacy in mice (17). In addition, we have shown that anti-DM4 PBSE administration was able to significantly increase mice tolerability of a DM4-based ADC, which enabled reduction in tumor volume to undetectable levels and to dramatic improvements in survival (24).
Antibody fragments are small in size, with high binding affinity and specificity, which makes them ideal as PBSEs. Leveraging the advancements in protein engineering technology, various antibody fragments, including Fab, sdAb, and scFv, can be generated in large quantities through various expression systems. The monoclonal antibody 8C2 has been extensively characterized in terms of its functional binding to topotecan and its ability to inhibit topotecan-induced toxicity in mice (20, 37). 8C2 Fab was selected as a candidate PBSE for the evaluation of our inverse targeting strategy on camptothecin derivative based ADCs. 8C2 IgG has been previously sequenced, allowing for the generation of Fab fragments through various approaches such as papain digestion and expression in E. coli or yeast systems. In this study, we opted for direct fragmentation from the intact IgG via papain digestion to obtain 8C2 Fab. Generation of antibody fragments in large quantities is often complicated by incomplete fragmentation of IgG, protein aggregation at high concentrations, and limited column binding capacity. To overcome these challenges, we employed a two-step purification method using protein G affinity chromatography, which is widely utilized for IgG purification due to its ability to bind both Fab and Fc fragments. Interestingly, our results showed that protein G binds more preferentially to 8C2 Fab fragments vs. Fc fragments. Similar results have been reported by Song et al. (38) during protein G purification of anti-myoglobin Fab fragments. Derrick and Wigley have identified an outer β-strand in the protein G domain III that forms an antiparallel interaction with the last β-strand in the Fab heavy chain CH1 domain, thereby extending the β-sheet structure and explaining the interaction mechanism between Fab and protein G (39).
The competitive MTT assay is a practical and cost-effective tool for screening of PBSE candidates for use for inverse targeting prior to in vivo evaluation. The assay is based on the premise that PBSEs present in the cell culture medium will bind to free drug and form extracellular complexes that are either too large or polar to diffuse across the cell membrane, thereby inhibiting drug-induced cell killing. Conversely, we hypothesize that PBSE binding to payloads in culture medium will not impact receptor-mediated endocytosis or internalization of ADCs, enabling the payload to be released intracellularly and exert its cytotoxic effect. Here we investigated the cell killing inhibition of PBSE on T-DXd and its payload DXd in HER2 overexpressing tumor cells. 8C2 Fab increased the DXd IC50 by > 50-fold, while there was no impact on T-DXd cytotoxicity. Our assay involved exposing cells to the medium containing T-DXd and 8C2 Fab for 24 h to allow binding between the ADC and cells and between the ADC and Fab. After this incubation period, drug-containing media was replaced with drug-free media. Consequently, the assay does not examine 8C2 Fab effects on the activity of DXd released into medium after 24 h (e.g., after cell lysis). The potential impacts of payload binding agents and payload in the tumor micro-environment in vivo and on cellular signaling pathways requires further investigation.
Modeling and simulation may provide valuable insights into the interaction between ADC, payload, and PBSE. Shah et al. previously published a hybrid PK model for topotecan with 8C2, which was used to predict the disposition of topotecan, 8C2, and the topotecan-8C2 complex (20, 21). A similar approach could be used for PBSE and ADCs and will be applied in future work.
Body weight loss was used in the present study to assess in vivo toxicity following dosing of T-DXd with or without 8C2 Fab. The advantages of using body weight loss as a toxicity marker include that it is non-invasive, easily measurable, and correlates strongly with the general health of rodents. In an intracranial rat glioma model, a fast deterioration in body weight was observed as a reliable predictor for clinical deterioration (40). Sewell et al. have shown an excess of 10% body weight loss in rats is highly predictive of death at higher drug concentrations, indicating the MTD had already been reached (41). Since there is no literature reported maximum tolerated dose (MTD) of T-DXd in mice (2), we conducted a single dose escalation study and identified a MTD of T-DXd of 600 mg/kg, which led to > 10% body weight loss at nadir. This dose level is greatly above the reported efficacious dose of T-DXd in HER2 tumor-bearing xenografts (42, 43). The translational assessment of toxicity for many ADCs under development, including T-DXd, is challenging as they are not cross-reactive with mice or rats. The low toxicity observed in the mice study may be attributed to the lack of target expression in mice and limited release of the payload into the systemic circulation. In addition, camptothecin derivatives have been reported to exhibit greater toxicity in humans compared to mice. For example, in vitro CFU-GM assays developed to predict myelotoxicity demonstrate that human cells are 2–20 fold more sensitive to CPT derivatives than mouse cells (6, 44, 45). The observed mouse body weight loss reached its maximum around day 3–4. In consideration of the time-course of absorption following IP dosing and the relatively short half-life of PBSEs compared to ADC, 8C2 Fab was administered in fractionated manner, with more frequent dosing during the first 24 h after T-DXd treatment, followed by dosing every 12 h (46). The 1:1 molar ratio of PBSE to payload in the ADC was selected empirically due to the unexpected high dose of ADC required to produce toxicity in mice. This ratio was deemed sufficient for proving the concept of PBSE mitigating ADC toxicity in vivo. The same molar ratio and fractional dosing regimen of 8C2 Fab was then employed in the T-DXd efficacy study. The result demonstrated that the anti-tumor effect of the ADC was not compromised by the presence of PBSE supporting our proposed inverse targeting strategy.
Despite the successful demonstration of the utility of the inverse targeting strategy using 8C2 Fab and T-DXd in our experiments, there is still room for optimization and further investigation. In future studies, optimal PBSE dosing regimens, including alternative routes of administration (e.g., subcutaneous dosing), will be explored through modeling and simulation. Available pharmacokinetic models for antibody fragments, small molecule agents, and ADC constructs can be integrated to evaluate the interaction between PBSE, payload, and ADC (20, 47, 48). Additionally, alternative toxicity models, such as cross-reactive rodent models or alternative toxicity biomarkers, such as hematological and clinical chemistry parameters, will be considered. The current efficacy model also lacks a full understanding of the bystander effect of the ADC, and more sophisticated mouse models may be explored in future work (43). Finally, the effect of PBSE-payload complex on the elimination of the payload molecule, particularly through the renal route, should be investigated. These considerations can provide insights into the mechanisms of inverse targeting and guide the development of this strategy for other ADCs and their corresponding PBSEs.

Comments (0)