
Remember me
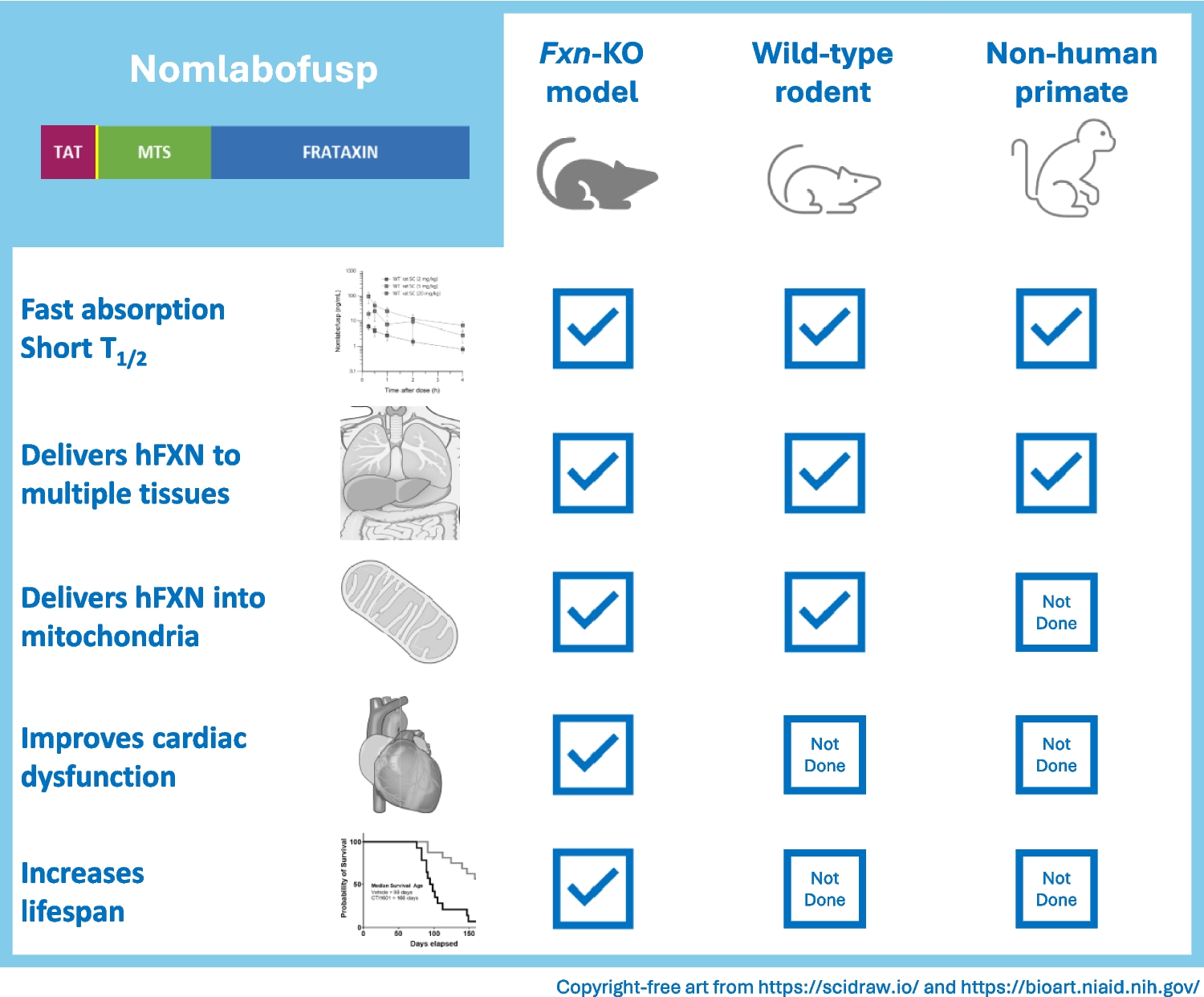
Following a single 10 mg/kg SC dose of nomlabofusp (equivalent to a 50 mg human dose) administered to C57BL6 WT mice and Fxn-KO mice, mean nomlabofusp concentrations were nearly indistinguishable between WT and KO. A higher dose of 50 mg/kg SC in the Fxn-KO mice also showed a similar PK profile, with fast SC absorption and elimination, and showed a dose-dependent increase in plasma concentrations (Fig. 1a). As seen in Table I, the 10 mg/kg and 50 mg/kg SC doses in the Fxn-KO mice produced approximately dose-proportional Cmax and AUC0-last. Nomlabofusp SC bioavailability was approximately 35%, as determined in WT mice.
Fig. 1
Nomlabofusp Plasma Pharmacokinetics in WT mice and rats, Fxn-KO mice and cynomolgus monkeys. Mean (SD) nomlabofusp plasma concentration–time profiles in a WT (C57BL6) mice after a single SC 10 mg/kg (n = 27 M/0 F) nomlabofusp dose and Fxn-KO mice after single SC 10 (n = 10 M/5 F) or 50 (n = 15 M/12 F) mg/kg nomlabofusp doses (n = 3/timepoint/group), samples were quantified using an electrochemiluminescence based immunoassay with an anti-TAT capture antibody; b WT Sprague Dawley rats treated daily SC with nomlabofusp 2, 5 or 20 mg/kg. PK samples were collected after 6 days of treatment (n = 9 M/timepoint/group), samples were quantified using hybrid LC–MS/MS that is specific to nomlabofusp (anti-TAT antibody capture); c Cynomolgus monkeys after a single SC 15 mg/kg nomlabofusp dose (n = 3 M/3 F); plasma samples were collected after the first dose and quantified using a qualified ELISA with anti-TAT antibody capture and anti-FXN antibody detection
Table I Summary of nomlabofusp plasma pharmacokinetics in WT and Fxn-KO mice. Wild-type (WT) C57BL6 mice were administered a single IV Dose of 5 mg/kg (n = 24 M/0 F) or single SC dose of 10 mg/kg (n = 27 M/0 F) nomlabofusp. Fxn-KO mice were administered a single SC dose of either 10 (n = 10 M/5 F) or 50 (n = 15 M/12 F) mg/kg Nomlabofusp (n = 3/timepoint/group). Plasma samples were collected at 0.083, 0.25, 0.5, 0.75, 1, 2, 4 and 7 h (IV and SC), and 24 h (SC only) post-dose. Samples were quantified using an electrochemiluminescence based immunoassay. PK parameters were calculated using the mean concentrations at each time point for AUClast, area under the plasma concentration Versus time curve to the last quantifiable time point; Tmax, time to maximum concentration; Cmax, maximum plasma concentrationSince PK parameters after SC administration of nomlabofusp in WT and KO mice were found to be comparable, we determined that studies in WT animals could be used to further evaluate the PK of nomlabofusp. Thereafter, dosing nomlabofusp daily at 2 mg/kg, 5 mg/kg, or 20 mg/kg in WT sprague dawley rats (equivalent to ~ 25, 50 and 225 mg in human) showed that it was rapidly absorbed after SC administration, with fast elimination and increasing exposure over this dose range (Fig. 1b). PK parameters calculated after the sixth dose are presented in Table II, where Mean Cmax and AUC0–last appeared dose-proportional.
Table II Summary of nomlabofusp plasma pharmacokinetics in WT rats. Wild-type (WT) Sprague Dawley rats (n = 17 M/group) were administered nomlabofusp SC at 2, 5 or 20 mg/kg daily for 7 days. Untreated Rats (n = 14 M) were used as controls. Plasma samples were collected on study day 6 at 0.25, 0.5, 1, 2 and 4 h post-dose and on day 7, 2.5 h post dose (Immediately Prior to Necropsy), and quantified using hybrid LC–MS/MS. Day 6 PK parameters were calculated using the mean profile. AUClast, area under the plasma concentration versutime curve to the last quantifiable time point; Tmax, time to maximum concentration; Cmax, maximum plasma concentrationLikewise, in healthy monkeys dosed with a single 15 mg/kg nomlabofusp SC dose, rapid absorption and elimination phases were observed (Fig. 1c).
Nomlabofusp Distributes to FRDA-relevant and Peripheral Tissues in Animal ModelsWT C57BL6 and Fxn-KO mice were administered a single SC dose of nomlabofusp at 10 mg/kg or 50 mg/kg. Brain, heart, liver, and skeletal muscle samples were harvested 1 h after dosing and flash frozen, which were later processed into tissue extracts. In all these samples hFXN concentration was quantified using a hybrid LC–MS/MS assay that excluded the detection of endogenous mouse FXN (see Supplementary File 2for assay schematics). As shown in Fig. 2a, hFXN concentrations were comparable between C57BL6 and Fxn-KO mice in all 4 tissues, with dose-dependent increase in hFXN after the 50 mg/kg nomlabofusp treatment in the Fxn-KO mice, supporting the use of WT animals to evaluate the pharmacodynamics (PD) of nomlabofusp. In the brain, hFXN was quantifiable in concentrations that were higher than the simultaneous plasma concentrations, leading to a brain-to-plasma concentration ratio > 0.04 (25), demonstrating hFXN penetration to brain above any potential contribution from blood contamination.
Fig. 2
Tissue levels of human FXN after subcutaneous administration of nomlabofusp. a Brain, heart, liver and skeletal muscle were harvested 1 h after a single 10 mg/kg SC nomlabofusp dose to wild-type (WT) C57BL6 mice (n = 3 M/0 F), a single 10 (n = 2 M/1 F) or 50 (n = 2 M/1 F) mg/kg dose of nomlabofusp to Fxn-KO mice. The tissues were frozen at −80°C and homogenized in RIPA buffer. FXN was quantified using hybrid LC–MS/MS that is specific to nomlabofusp (anti-TAT antibody capture). Peptide SGT was used for detection. Results were normalized to tissue mass. Mean and SD are shown; black circles represent data points from individual animals. b Wild-type (WT) Sprague Dawley rats (n = 12 M/group) were administered 2, 5 or 20 mg/kg nomlabofusp daily for 7 days via SC injection. The untreated group (n = 14 M, dose = 0 mg/kg) remained on study for the same duration as the nomlabofusp treated groups. Approximately 2.5 h after the 7 th dose, half of the animals in each dose group were perfused with PBS and tissues harvested. FXN was quantified using hybrid LC–MS/MS (anti-FXN antibody capture). Peptide SGT was used for detection. Results were normalized to the total tissue protein content of the sample and expressed in pg/μg. Median tissue concentrations and IQR for hFXN or endogenous (rat) FXN in unperfused (n = 6 M/dose group) and perfused (n = 6 M/dose group) animals in the dorsal root ganglion, skeletal muscle, liver, cerebellum, cerebrum and skin are shown. Black circles represent data points from individual animals. c Healthy cynomolgus monkeys were administered 15 mg/kg nomlabofusp twice daily for 14 days starting on Study Day 3. Each animal served as its own control by receiving vehicle for two days before receiving nomlabofusp. On Study Days 10 and 16, tissue collections included whole blood for platelets, skin punch biopsies and buccal swabs. Tissue and cellular homogenates were prepared and hFXN determined using hybrid LC–MS/MS (anti-FXN antibody capture). Peptides SGT and LGG were used for detection. Results were normalized to the total tissue protein content of the sample and expressed in pg/μg. Human FXN values in buccal (n = 3 M/3 F), platelets (n = 3 M/3 F) and skin (n = 3 M/3 F) from the vehicle treated monkeys were below the level of quantification (BLQ). Mean and SD are shown. Refer to Supplementary File 2for assay schematics (specific peptides)
To understand nomlabofusp exposure and FXN distribution in tissues after multiple doses, WT Sprague Dawley rats were administered 2 mg/kg, 5 mg/kg, or 20 mg/kg nomlabofusp SC daily for 7 days. After the final dose the cerebellum, cerebrum, DRG, liver, heart, skeletal muscle, and skin were collected and tissue homogenates prepared. Endogenous rat FXN and hFXN were quantified by a hybrid LC–MS/MS assay capable of differentiating species-specific FXN. In this experiment, whole-body perfused rats (prior to tissue harvest) were also included to ensure that the observed tissue concentrations would exclude contamination by blood. As seen in Fig. 2b, perfusion had no effect on the endogenous levels of FXN, as would be expected. Across doses in all the rats, the median rat FXN concentrations were 19.3, 60.5, 70.0, 12.2, 17.1, 44.7, and 39.1 pg/μg in perfused DRG, heart, liver, skeletal muscle, skin, cerebellum, and cerebrum, respectively, indicating that endogenous FXN levels are tissue specific. In unperfused rats, nomlabofusp-derived hFXN was measurable in these tissues, showing a dose-dependent increase. Also, in tissues from unperfused rats hFXN concentrations were higher compared with DRG, heart and liver from perfused rats and were not quantifiable in the cerebrum and cerebellum of perfused rats. In skeletal muscle and skin negligible differences were observed in perfused versus unperfused samples.
Significant correlations between hFXN in the tissues were also observed, as shown in Fig. 3 (heart/DRG, r = 0.76; skeletal muscle/DRG, r = 0.58; heart/skeletal muscle, r = 0.49). Notably, hFXN increases after nomlabofusp administration in these primary target organs correlated with hFXN increases in the skin, a peripheral tissue (skin/heart, r = 0.82; skin/DRG, r = 0.84; skin/skeletal muscle, r = 0.66). Skin showed the greatest hFXN increase in 7 days (Fig. 2b) and it correlated with the hFXN levels in other organs (Fig. 3), suggesting that skin is a predictor of nomlabofusp penetration into FRDA-relevant tissues. A tissue plasma correlation was also observed (see Supplementary File 5), where hFXN increases in tissues correlated with nomlabofusp concentration in plasma at the time of tissue collections.
Fig. 3
Tissue levels of human FXN amongst various tissues after subcutaneous administration of nomlabofusp in rats. WT rats (n = 12 M/group) were dosed SC with 2, 5 or 20 mg/kg nomlabofusp daily for 7 days. Approximately 2.5 h after the 7 th dose, half of each dose group (n = 6 M) were perfused and organs harvested. Human FXN in each tissue was quantified using hybrid LC–MS/MS (anti-FXN antibody capture, peptide SGT for detection).and was normalized to the total tissue protein content of the sample and expressed in pg/μg. Comparisons of the concentrations of hFXN in various perfused tissues are shown with Pearson correlation coefficient (Corr) and significance (p). Refer to Supplementary File 2 for assay schematics (specific peptides)
A longer multiple dose study was conducted in healthy monkeys in which nomlabofusp was administered twice a day SC at 15 mg/kg daily for 14 days (Study Days 3 through 16). Each monkey served as its own control by receiving vehicle alone for 2 days prior to receiving nomlabofusp (Study Days 1 and 2). Tissue samples (buccal swab, skin biopsy, and platelets) were collected on Days 3 (pre-dose), 10, and 16 of nomlabofusp treatment and processed into homogenates in which hFXN, monkey FXN, and nomlabofusp were quantified using hybrid LC–MS/MS. Endogenous monkey FXN was quantifiable in all pre-dose samples. As shown in Fig. 2c, hFXN was present in buccal cells, platelets, and skin as early as Day 10 and had similar levels on Day 16. A small fraction of nomlabofusp was detected in buccal (< 25%) and skin (< 5%) but not in platelets compared to hFXN (see Supplementary File 6). Using a nomlabofusp-specific ELISA, nomlabofusp was not detected in any of the CSF samples. However, in the same monkeys, CSF, collected at pre-dose (Study Day 3) and at necropsy (Study Day 16), was tested with another ELISA that detected immunoreactive hFXN (present in both hFXN and nomlabofusp). With this method, signal was quantifiable in all post-treatment samples at concentrations ranging from 1.31 to 5.52 ng/mL, indicating that hFXN was present in the CSF (see Supplementary File 6).
Nomlabofusp Produces Dose-dependent Increases of hFXN in Mitochondria and is Processed to Mature hFXN in RodentsIn the WT and Fxn-KO mice that were administered a single SC dose of nomlabofusp at 10 mg/kg or 50 mg/kg, liver mitochondria were harvested 1 h after dosing and processed to obtain ME. Human FXN was quantified using a hybrid LC–MS/MS assay. As shown in Fig. 4a, hFXN concentrations in liver ME were comparable between C57BL6 and Fxn-KO mice and had a dose-dependent increase.
Fig. 4
Human FXN in mitochondria after subcutaneous administration of nomlabofusp. a Liver was harvested 1 h after a single 10 mg/kg SC nomlabofusp dose to wild-type (WT) C57BL6 mice (n = 3 M/0 F), a single 10 (n = 2 M/1 F) or 50 (n = 2 M/1 F) mg/kg dose of nomlabofusp to Fxn-KO mice. Liver mitochondria extracts (“Liver ME”) were prepared on day of harvest using fresh tissue. Human FXN concentrations were quantified using hybrid LC–MS/MS (anti-FXN antibody capture, peptide SGT for detection). Liver ME hFXN concentrations were normalized to the total tissue protein content measured using a BCA assay (ng/mg). Mean and SD are shown; black circles represent data points from individual animals. Refer to Supplementary File 2 for assay schematics (specific peptides). b WT rats (n = 5 M/group) were administered 20 mg/kg nomlabofusp daily for 7 days via SC injection. Approximately 2.5 h after the 7 th dose animals were perfused with PBS and tissues harvested. Mitochondria were isolated from the heart and skeletal muscle. All samples from each tissue were pooled. FXN was immunoprecipitated from 500 µg total protein from heart mitochondria and 431 µg total protein from skeletal muscle mitochondria using a human-specific FXN antibody and analyzed by western blotting. Nomlabofusp and HEK293 cell extracts were immunoprecipitated as controls to identify the migration of full length nomlabofusp and mature hFXN, respectively
Having demonstrated that nomlabofusp delivered hFXN into liver mitochondria, it became pertinent to determine whether intramitochondrial hFXN had been processed into mature hFXN within FRDA-relevant tissues. From whole-body perfused rats dosed with 20 mg/kg nomlabofusp, heart and skeletal muscle whole cell homogenates were used to isolate mitochondria, which were then lysed and their extracts collected. For each tissue, ME samples from all rats were pooled and FXN was immunoprecipitated using a hFXN-specific antibody, followed by SDS-PAGE based separation and western blot analysis (Fig. 4b). The band between 25–30 kDa is where nomlabofusp migrates on SDS-PAGE. HEK293 cells demonstrate the presence of mature hFXN (14.3 kDa), as well as endogenous immature/unprocessed hFXN (23.1 kDa), which migrates with nomlabofusp on the blot due to their minor molecular weight difference. The presence of 14.3 kDa bands in heart and skeletal muscle ME were consistent with processed, mature FXN. This result in animals confirms previously published findings, where the presence of mature hFXN was demonstrated in nomlabofusp exposed cell lines and buccal tissue of nomlabofusp dosed patients with FRDA (19). Interestingly, a band consistent with the ~ 17 kDa intermediate form (FXN56–210), was also clearly visible in the heart, and less so in skeletal muscle.
Nomlabofusp Restores SDH Activity in Fxn-KO MiceFxn-KO mice were administered nomlabofusp at 5 or 6 weeks of age, when the impact of FXN deficiency had begun to manifest via early signs of progressive cardiomyopathy. As shown in Fig. 5a, nomlabofusp significantly increased SDH activity in heart mitochondria of the Fxn-KO mice when dosed every other day for 14 days starting at 5 weeks of age or daily for 20 days starting at 6 weeks of age. Dose-dependent increases were evident between 0.4 mg/kg and 2 mg/kg (p < 0.0001) and between the 2 mg/kg and 10 mg/kg dose groups. When mice began receiving nomlabofusp at the age of 6 weeks (Fig. 5b), the 0.4 and 2 mg/kg groups were not statistically different than vehicle treated Fxn-KO mice, but the 10 mg/kg/day dose resulted in a significant (p < 0.02) increase in SDH activity compared to vehicle treated Fxn-KO mice.
Fig. 5
SDH activity after SC administration of nomlabofusp in WT and Fxn-KO mice. a Heart: Starting at 5 weeks of age Fxn-KO mice were administered nomlabofusp at 0.4 (n = 4 M/4 F), 2 (n = 3 M/4 F) or 10 (n = 1 M) mg/kg SC every 48 h for 14 days. After the last dose hearts were harvested at necropsy, and mitochondrial extracts were prepared. SDH activity was measured in heart mitochondrial extracts and mean and SD are shown; black circles represent data points from individual animals. Two-way ANOVA was performed. b Heart: Starting at 6 weeks of age Fxn-KO mice were administered nomlabofusp at 0.4 (n = 2 M/2 F), 2 (n = 2 M/1 F), 10 (n = 2 M/2 F) mg/kg or vehicle SC daily (n = 2 M/2 F) for 20 days. After the last dose hearts were harvested at necropsy, and mitochondrial extracts were prepared. SDH activity was measured in heart mitochondrial extracts and mean and SD are shown; black circles represent data points from individual animals. c Skeletal Muscle: Fxn-KO mice were treated SC every 48 h for 14 days with 2, 10, 30, 60 or 100 mg/kg (n = 4 M/4 F/group) nomlabofusp or vehicle (n = 4 M/4 F). WT mice (n = 6 M/6 F) were treated SC every 48 h for 14 days with vehicle. After the last dose, skeletal muscle tissue was harvested at necropsy and mitochondrial extracts were prepared. SDH activity was measured in skeletal muscle mitochondrial extracts and mean and SD are shown; black circles represent data points from individual animals. Two-way ANOVA was performed with post-hoc (Tukey) comparison of the parameters
Nomlabofusp significantly increased SDH activity in skeletal muscle mitochondria of the Fxn-KO treated mice when dosed every other day for 14 days starting at 5 weeks of age (Fig. 5c). The 2 mg/kg dose seemed ineffective, whereas a notable but not statistically significant increase was seen at 10 mg/kg (p = 0.49). Nevertheless, significant (p < 0.01) increases relative to vehicle treated Fxn-KO mice were evident at 30 mg/kg and 100 mg/kg, reaching levels that appeared comparable to WT mice suggesting a plateau effect had been reached.
Nomlabofusp Treatment Halts Cardiac Dysfunction in Fxn-KO MiceFxn-KO or WT mice were treated SC with 10 mg/kg nomlabofusp or vehicle every other day for 6 weeks, starting at 5 weeks through 11 weeks of age. Baseline cardiac performance was determined by echocardiography at 4 weeks of age prior to the first dose, and post-treatment assessment was done after the mice were 8 weeks of age. Imaging data are shown from representative mice in Fig. 6a. The green demarcated areas represent the left ventricular walls for strain analysis in the vehicle and nomlabofusp treated mice at 8 weeks of age, respectively.
Fig. 6
Cardiac function in WT and Fxn-KO mice after subcutaneous administration of nomlabofusp. WT or 5-week-old Fxn-KO (n = 4 M/4 F/group) were treated with either vehicle or nomlabofusp SC at 10 mg/kg every 48 h for 6 weeks. Ultrasound echocardiography assessments were completed at 4 (pre-dose) and 8 weeks of age. a Heart echocardiograph images were captured at 8 weeks of age and ejection fraction and global longitudinal strain quantified. Cardiac echocardiography parameters were determined: b Left Ventricle Ejection Fraction (EF, %), c Fractional Shortening (FS, %), d Left Ventricular Cardiac Output (CO, mL/min), e Left Ventricle Stroke Volume (SV, µl), f Left Ventricle Internal Dimension (D;s, mm), and g Left Ventricle Volume, systole (V;s, µl). Mean and SD are shown. Initiation of dosing at 5 weeks of age is denoted by a caret (^). Two-way ANOVA was performed with post-hoc (Tukey) comparison of the parameters. Statistical differences between vehicle treated WT and Fxn-KO mice or nomlabofusp treated Fxn-KO mice and vehicle treated WT mice are shown
In Fxn-KO mice disease onset was demonstrated at 4 weeks of age by the presence of compromised cardiac function measured by 4 parameters that were significantly different compared with WT mice: ejection fraction (p < 0.002), fractional shortening (p < 0.007), cardiac output (p < 0.0001) and stroke volume (p < 0.0008), as shown in Fig. 6b-e. In vehicle treated Fxn-KO mice cardiac function continued to decline as the mice aged, with further worsening at 8 weeks of age in all 4 parameters observed with statistically significant differences versus WT: ejection fraction (p < 0.001), fractional shortening (p < 0.0001), cardiac output (p < 0.01), and stroke volume (p < 0.01) (Fig. 6b-e). In contrast, in the Fxn-KO mice treated with nomlabofusp these parameters did not decline further by 8 weeks of age and were comparable to WT vehicle treated mice (Fig. 6b-e; statistically insignificant difference with any of the cardiac functional parameters measured). At 4 weeks of age, the decline in cardiac functional output between the study groups was not predicted by alterations in left ventricle size, as neither left ventricle interior dimension (LVID) nor left ventricle volume (LV Vol) were significantly different (Fig. 6f and g). However, by 8 weeks of age, the vehicle treated Fxn-KO mice demonstrated significant differences in both left ventricle parameters when compared to vehicle treated WT mice. Left ventricular internal diameter was significantly increased (p = 0.0001), as was LV Vol (p < 0.001) when compared to WT mice (Fig. 6f and g). Importantly, nomlabofusp treatment prevented the decline in left ventricular parameters, resulting in no significant difference between WT and treated Fxn-KO mice by 8 weeks of age (Fig. 6f and g).
Collectively, the data in Fig. 6 demonstrated that delivering exogenous FXN via nomlabofusp administration in these mice prevented further decline in cardiac function and for some outcome measures, such as ejection fraction, the function was comparable to age-matched vehicle treated WT mice.
Nomlabofusp Treatment Leads to Improved Survival of Fxn-KO MiceAs shown in Fig. 7, Fxn-KO mice treated SC with nomlabofusp, 10 mg/kg every other day starting at 2 weeks of age, lived significantly longer than mice treated with vehicle (log rank analysis, p < 0.0001). The median survival for mice treated with nomlabofusp was 166 days, compared to 98 days for vehicle-treated mice. At the end of the study period (170 days), none of the vehicle treated mice (n = 15) were alive while 8 of the 16 nomlabofusp treated mice were alive. Of the 8 that remained alive, 3 were male and 5 were female, indicating that there was no evidence of sex difference in survival.
Fig. 7
Survival in Fxn-KO mice after treatment with nomlabofusp. Fxn-KO mice entered the study at 14–16 days of age and were treated with 10 mg/kg nomlabofusp (n = 8 M/8 F) or vehicle (n = 8 M/8 F) SC 3 times per week up to 170 days of life, until they died, or were removed from the study by 200 days. Data were analyzed by Kaplan–Meier Survival curve with log rank analysis (p = 0.0001)
Comments (0)