2.1 Data Collection
Children were retrospectively enrolled from a tertiary pediatric hospital center (CHU Sainte-Justine, Montreal, QC, Canada). Inclusion criteria included SOT or HSCT transplantation between January 2007 and December 2015, receiving valganciclovir and/or intravenous ganciclovir for CMV disease prevention, an available pharmacokinetic profile, and at least two CMV viral loads per patient. Patients who received foscarnet were excluded from the analysis. At CHU Sainte-Justine, the CMV disease prevention strategy involves a pre-emptive approach: CMV DNA in peripheral blood is monitored weekly for the initial 100 days after the transplant and then monthly for up to 6 months. Generally, antiviral therapy with intravenous ganciclovir 5 mg/kg every 12 h (q12 h) or enteral valganciclovir 10 mg/kg q12 h is initiated upon detection of CMV DNA exceeding a significant threshold, which is not standardized and varies based on risk factors (CMV DNAemia level, time since transplant, CMV status). The dosing regimen can be adjusted according to the clinical condition of the patient (renal status, TDM); for example, some patients may receive (val)ganciclovir q24 h instead of q12 h. To ensure consistency across the dataset, AUC values from two patients who initially received q12 h dosing and were later switched to q24 h dosing were divided by two, allowing all data to be expressed in a q12 h framework. Repeated CMV episodes were considered as independent events if they occurred after treatment discontinuation of at least 3 months, reflecting clinically distinct recurrences rather than breakthrough infections. The institutional review board of the CHU Sainte-Justine approved the protocol (2018-1830).
2.2 Sampling and Bioanalysis
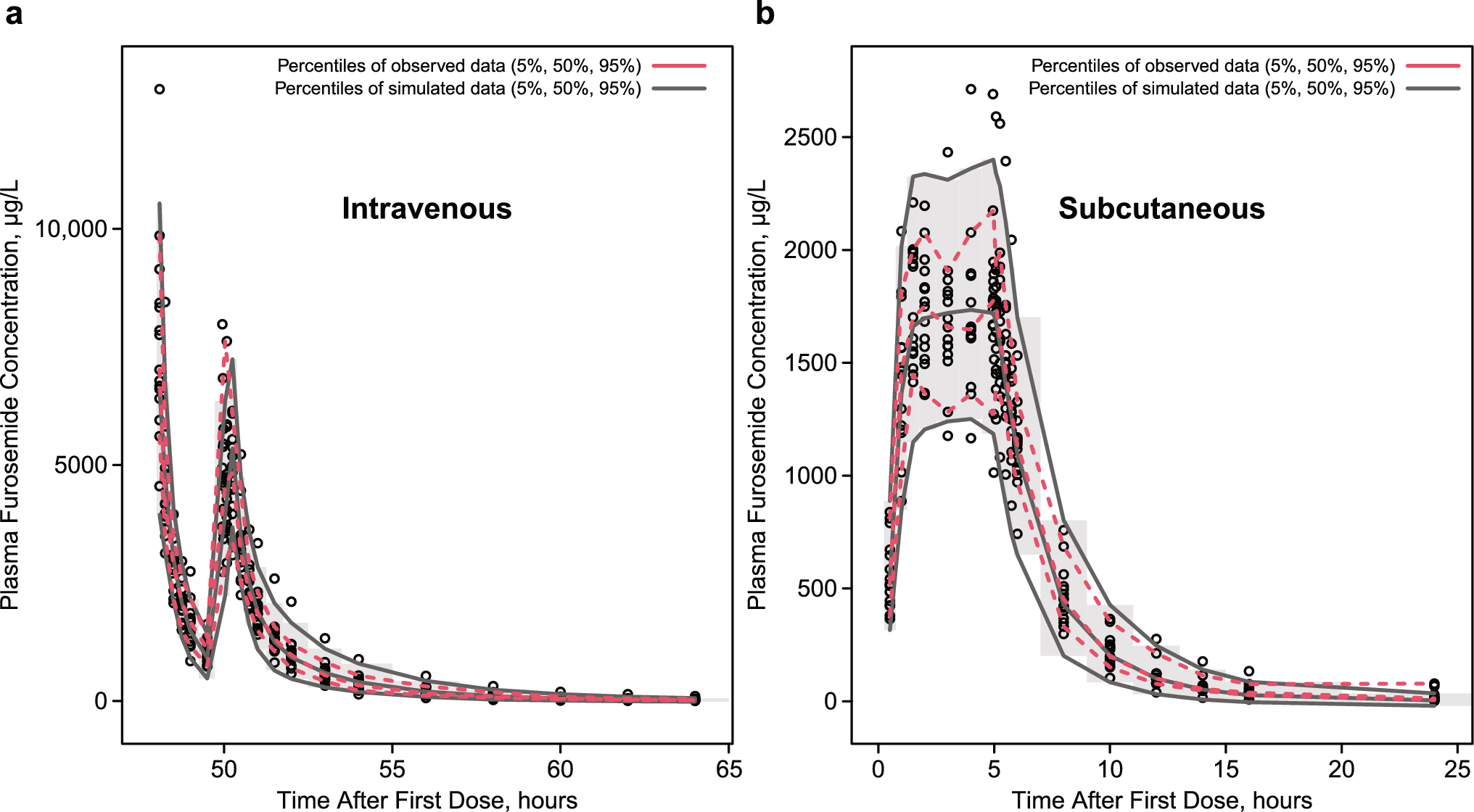
In accordance with local standards of care, TDM of ganciclovir was initiated after at least six doses (72 h) to reach steady state (half-life 4–10 h), with a target AUC0–24 range of 40–60 mg·h/L (derived from adult prophylaxis data). Blood samples were collected routinely in EDTA microtainers (0.25 mL) at specified time points: pre-dose and 0.5, 1, 1.5, 2, 3, 6, and 12 h for intravenous ganciclovir, and pre-dose and 0.5, 0.75, 1, 1.5, 2, 4, 6, and 12 h for enteral valganciclovir. Samples were immediately centrifuged at 3500 rpm for 10 min (2634 g) and stored at −30 °C in the hospital clinical laboratory. Plasma concentrations were analyzed within 72 h of sampling using high-performance liquid chromatography with diode array detection [15]. CMV viral loads were determined using a custom-developed quantitative polymerase chain reaction assay, which utilizes a standard curve derived from quantified commercial controls sourced from Advanced Biotechnology (strain AD169) with a lower limit of quantification (LLOQ) at 200 copies/mL. All aspects of the ganciclovir assay (including accuracy, precision, and the LOQ) have been rigorously validated in accordance with established guidelines.
2.3 Pharmacokinetic and Pharmacodynamic Modelling
The model was developed using a non-linear mixed effect approach and the stochastic approximation expectation-maximization algorithm with the software Monolix (version 2023R1, Lixoft SAS, a Simulation plus company). Pharmacokinetics and pharmacodynamics were modeled sequentially. First, we used a previously published two compartmental pharmacokinetic model with double gamma absorption to estimate individual AUC0–12 [13, 16]. This model was developed with an iterative two-stage Bayesian method with the same dataset as the current study. Original parameter ranges were reinterpreted as 95% confidence intervals of lognormal distributions. The fraction parameter r was obtained similarly but assuming a logit distribution instead. We estimated the AUC12 for each patient based on the empirical Bayesian estimates.
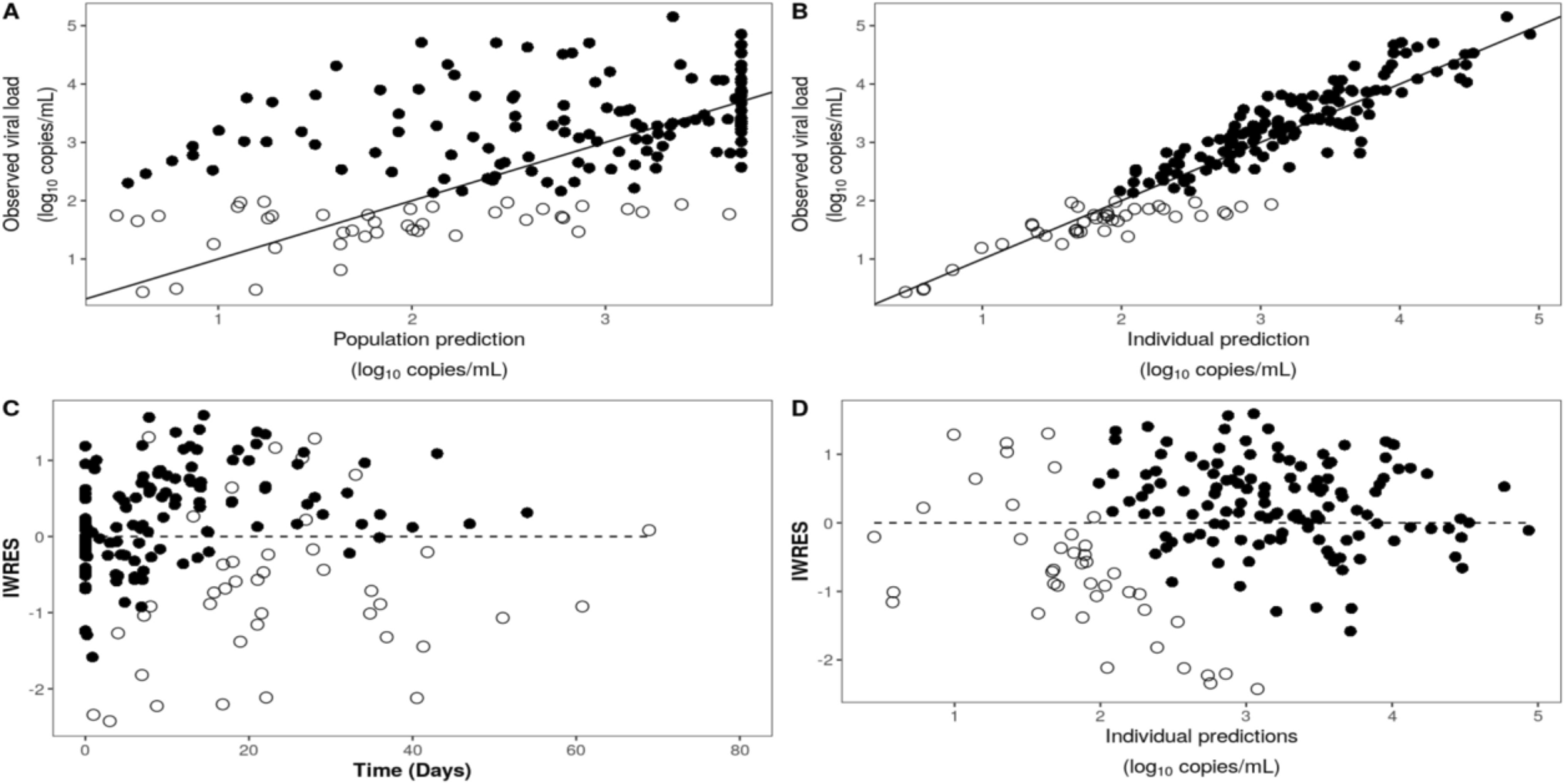
We then modeled the CMV viral loads with an indirect viral turnover model with stimulation of the viral degradation. Pharmacodynamic data below the LOQ were included in the analysis according to the NONMEM M4 method [17]. This method has been shown to perform well in a previous PK/PD model for (val)ganciclovir and CMV viral loads in adult kidney transplant recipients [18]. To assess the relationship between AUC0–12 and viral loads, we computed AUCs with the pharmacokinetic model and put them into the pharmacodynamic model, as in Eq. 1.
$$\frac= _- _ \times \left(1+\frac_*}_}_+}_}\right) \times \text$$
(1)
where R denotes the response, (i.e. the CMV viral load in plasma), \(\frac\) signifies the rate of change of viral load in plasma over time, \(_\) and \(_\) denote the rates of increase and decrease in CMV viral load in plasma, respectively, \(_\) indicates the ganciclovir concentration necessary for half-maximal rate of inactivation, and \(_\) represents the maximum rate of decline in CMV viral load. The initial CMV viral load at time zero (R0) equates to the ratio \(\frac_}_}\).
AUC0–12 was implemented as a time-dependent covariate in the pharmacodynamic model by integrating individual predicted AUC values from the pharmacokinetic model as a varying input (‘amount’) in Monolix, thus capturing intra-individual changes due to TDM-based dose adjustments and regimen modifications, including shifts from q12 h to q24 h dosing.
Type of transplant (SOT or HSCT), time since transplantation, and graft versus host disease (GVHD) were examined as covariates to determine whether they correlated with pharmacodynamic parameters. Of note, the potential impact of GVHD was tested both as a binary covariate (yes vs. no) and as a time-dependent variable, based on the documented onset of GVHD and its treatment. Pharmacodynamic covariate analyses were made using a forward inclusion (p < 0.05) and a backward deletion (p < 0.01) procedure.
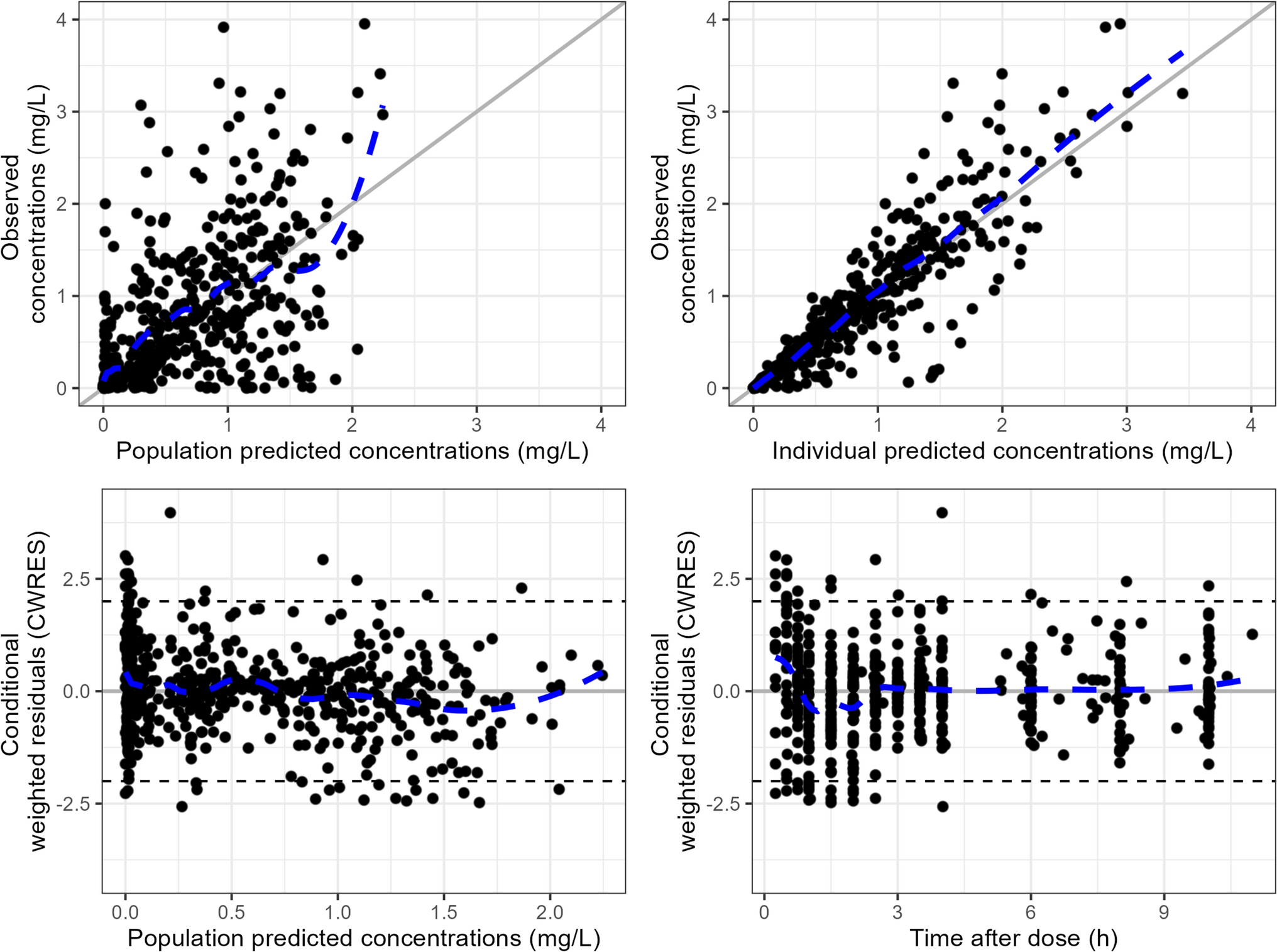
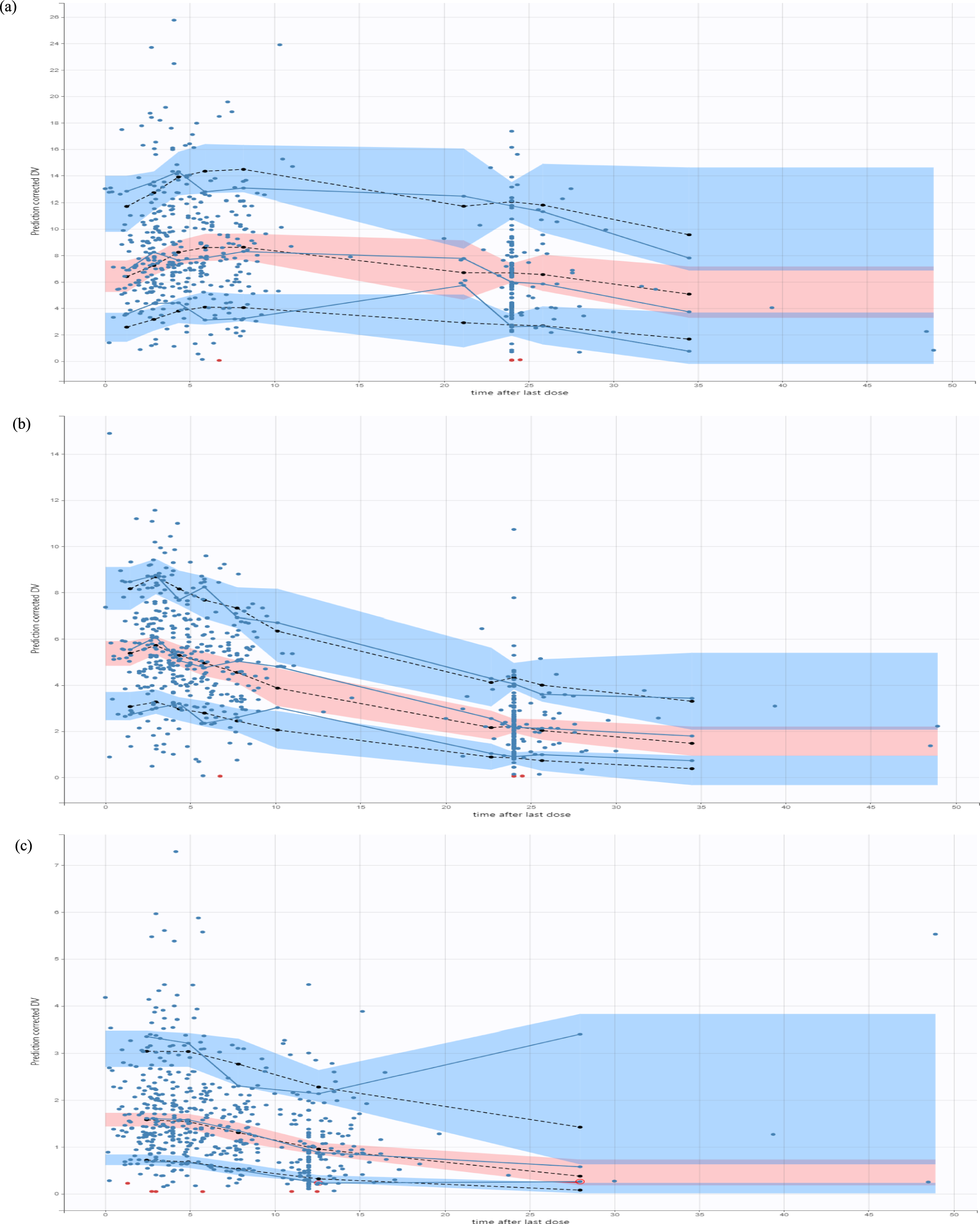
Internal validation was performed using goodness-of-fit plots, visual predictive checks, and 100 bootstraps. All plots were drawn in R (version 4.2.2).
2.4 Monte Carlo Simulations
Monte Carlo simulations were performed using Simulx (version 2023R1, Lixoft SAS, a Simulation plus company). Simulations were conducted using AUC0–24, in line with clinical exposure targets, assuming AUC0–24 ≈ 2 × AUC0–12 under steady-state conditions. Final estimates, including inter-individual variability, of the pharmacodynamic model were used to generate 1000 CMV viral loads versus time profiles with various AUC0–24 from 10 mg.h/L to 80 mg.h/L (same individual among groups). We categorized the simulated CMV viral loads into detectable or undetectable groups, using a threshold of 2 log10 copies/mL, determined by the LOQ of our CMV DNA monitoring assay. The probability of a decrease of 1 log10 copies/mL after 2 weeks of treatment according to The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation [2] was assessed. According to the Guidelines of the American Society of Transplantation Infectious Disease Community of Practice [19], antiviral therapy should be continued until DNAemia is no longer detectable, so we also evaluated the probability of unquantifiable viral load within a month of treatment. We were then able to plot the probability of achieving undetectable viral loads over time across different AUC0–24 values.


Comments (0)