
Remember me
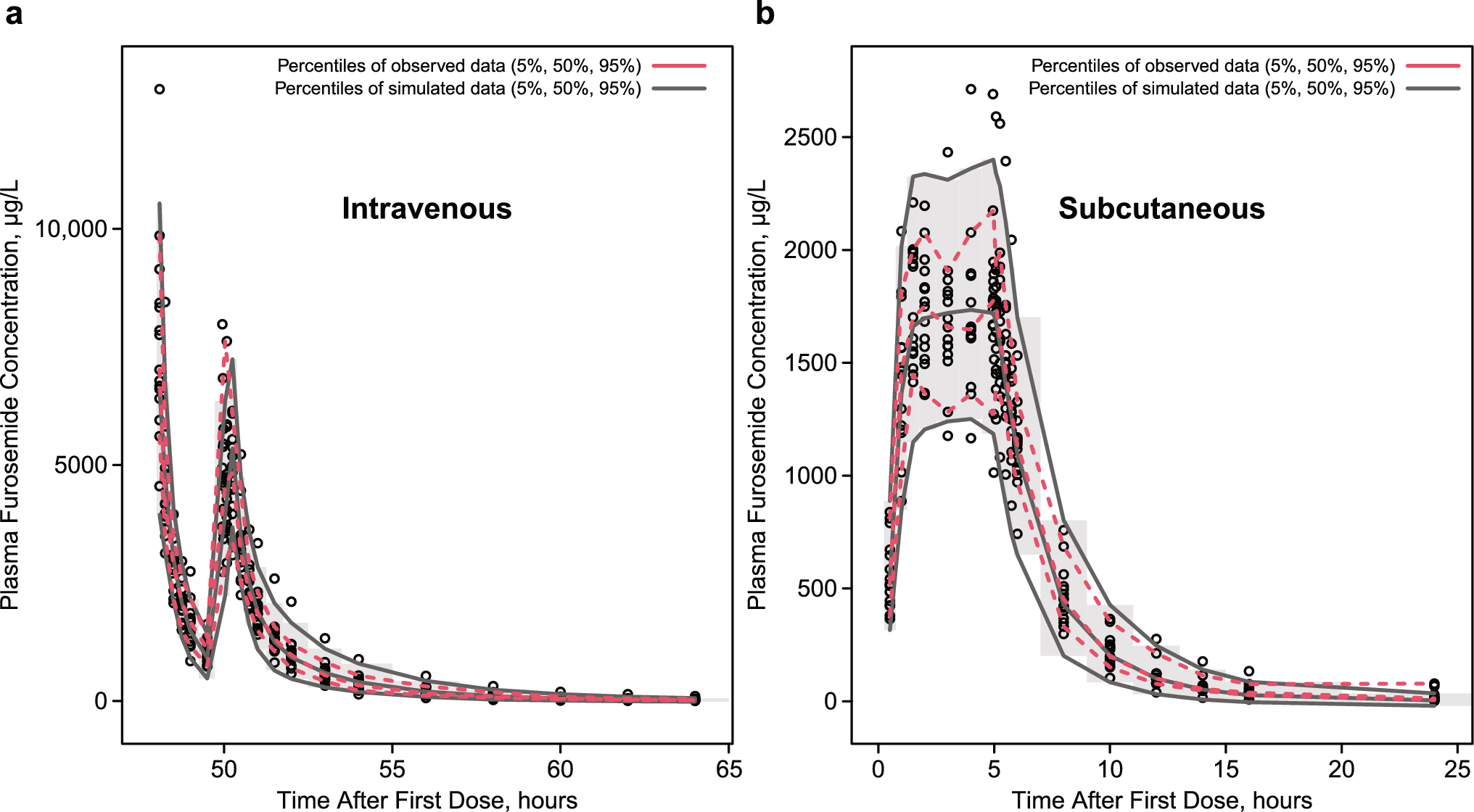

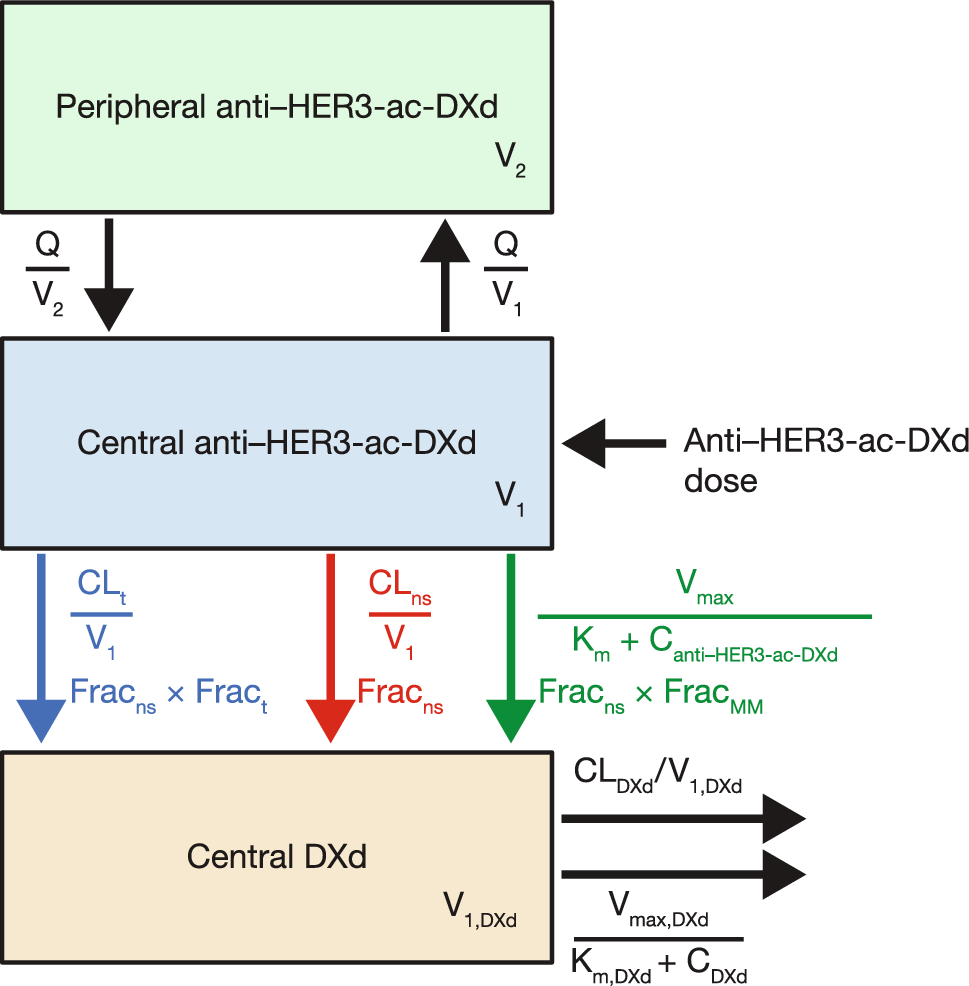
Diuretics are mainly classified by their site of action within the nephron, which closely relates to their efficacy. Except for spironolactone and its analogs, all diuretics act on the luminal side of tubular cells. Their secretion into the tubular lumen involves distinct transport mechanisms: loop diuretics (LDs) and thiazides rely ontorganic anion Transporters (OATs) and multidrug resistance-associated protein 4, while amiloride and triamterene use organic cation transporters (OCTs). LDs and hydrochlorothiazide are taken up at the basolateral membrane via these transporters and delivered to the tubular fluid through apical transporters in a two-step transcellular process. A small amount of furosemide is also filtered through the glomerulus; however, owing to its high protein binding (> 98%), only a minimal fraction undergoes filtration. This strong plasma protein binding supports its tubular secretion and enhances its diuretic effect. In contrast, spironolactone and other aldosterone antagonists are not transported by organic transporters but instead act on cytosolic receptors. LDs inhibit the cotransport isoform 2 of sodium (Na+), potassium (K+), and chloride ions (Cl−) (NKCC-2) across the cell membrane along the ascending limb of the loop of Henle and the macula densa [6, 7] (Fig. 1). The NKCC-2 is electrically neutral and becomes activated when all four binding sites are occupied. Once Na+ enters the tubular cell from one side, it is subsequently extruded into the systemic circulation through the Na+/K+-ATPase-dependent transporter located in the basolateral membrane, thereby creating a favorable electrochemical gradient. For Na+ to enter the cell from the luminal side, where its concentration is lower, it relies on Na+/K+-ATPase pumps that actively expel Na+ from the opposite side of the tubular cell [6,7,8]. The luminal membrane of the thick segment of the loop of Henle exhibits high permeability to K+ owing to the presence of renal outer medullary potassium (ROMK) channels on the luminal membrane of the tubular cells, which serve as apical pathways for recycling potassium. On the basolateral side, chloride channels (CLCKb) facilitate the passage of Cl− across the basolateral membrane. At this stage, the potential difference across the membrane achieves a state of equilibrium, resulting in hyperpolarization for K+. Conversely, at the opposite pole of the cell, the basolateral membrane’s permeability to Cl− leads to depolarization. This combination of hyperpolarization of the luminal membrane and depolarization of the basolateral membrane gives rise to a potential difference of approximately 10 mV, with the luminal side being positive and the interstitial space being negative. This potential difference repels divalent cations such as Ca2+ and Mg2+; however, it also serves as an important driving force for their transport via paracellin-1 through the paracellular route [7, 8].
Fig. 1
The mechanism of action of the Loop diuretics. ROMK renal outer medullary potassium
The loop of Henle is virtually impermeable to water, as it lacks aquaporins in contrast to other segments of the proximal tubule. Consequently, water is not reabsorbed at this level, while sodium and chloride are symported, leading to a dilution of tubular fluid by 25–35% owing to the removal of Na+ and Cl− [6, 8]. If the ion symport mechanism is blocked by LDs, this percentage of Na+ is not reabsorbed, remaining in the tubular fluid as it progresses to the collecting duct. There, it may be reabsorbed to a limited extent under the influence of aldosterone, eliminating K+. The Na+ that remains in the tubular fluid osmotically drags water along with it, which accounts for the diuretic effect of LD as well as side effects such as hyponatremia, hypokalemia, hypocalcemia, and hypomagnesemia [7, 8].
Several inhibitors of the NKCC-2 symporter exhibit additional effects in the proximal tubule. For instance, furosemide possesses weak carbonic anhydrase-inhibiting activity, resulting in increased urinary excretion of bicarbonate (HCO3−) and phosphate. In contrast, EA and bumetanide lack carbonic anhydrase-inhibiting activity [9].
3.2 Pharmacokinetics of Loop DiureticsTable 1 shows the pharmacokinetic characteristics of LDs. Furosemide, bumetanide, EA, and torsemide are rapidly absorbed following oral administration, achieving peak concentrations (Cmax) approximately 30 to 120 min postingestion. In contrast, when administered intravenously, the effects of furosemide and EA are nearly immediate [8,9,10].
Table 1 The pharmacokinetic characteristics of loop diureticsThe bioavailability of torsemide and bumetanide after oral administration is around 80%, while EA has an approximate bioavailability of 100%. Conversely, furosemide’s bioavailability is considerably lower and highly variable, ranging from 10% to 90%, and is significantly influenced by concurrent food intake. Consequently, when transitioning from intravenous to oral administration, the doses of torsemide and bumetanide should remain unchanged, whereas the dose of furosemide should be doubled to account for its variable absorption [8, 10].
The half-life (t½) of Furosemide is relatively short, at approximately 90 min; however, its duration of action can extend up to about 6 h following oral administration. This extended duration is likely due to gastrointestinal absorption occurring at a rate slower than its half-life, a phenomenon referred to as “absorption limited kinetics.” This limitation does not apply to torsemide and bumetanide, both of which exhibit rapid oral absorption. Indeed, although food slows and lowers the peak plasma concentration of bumetanide, it does not significantly affect the total amount absorbed or excreted, indicating only a minor impact on its overall bioavailability—especially when compared with furosemide [11]. Similarly, coadministration of torsemide with a high-fat, high-carbohydrate meal reduces its absorption rate without altering the extent of absorption, half-life, or renal clearance. These modest pharmacokinetic changes do not influence the drug’s diuretic efficacy, as the relationship between urinary sodium excretion and drug levels, as well as total electrolyte and urine output, remain unaffected [12].
EA has a half-life of approximately 30 min, which can vary from 12 to 160 min following intravenous administration [8, 10]. Similar to furosemide, the diuretic effect of EA is relatively short-lived. The drug is efficiently metabolized and eliminated by both the liver and kidneys, thereby preventing accumulation, even with multiple doses [9].
Approximately 40% of a furosemide dose is excreted unchanged in the urine and undergoes metabolic modification through glucuronidation in the kidneys. In contrast, torsemide and bumetanide are primarily eliminated via the liver before urinary excretion. For furosemide, metabolic variations among patients can influence the drug’s half-life, particularly in individuals with acute kidney injury (AKI) or chronic kidney disease (CKD), where renal excretion and glucuronidation may be impaired. Conversely, the half-lives of torsemide and bumetanide remain relatively stable in patients with renal dysfunction. After intravenous administration of EA, approximately one-third is excreted by the liver, while two-thirds are eliminated by the kidneys. The drug recovered from the urine is roughly distributed into three fractions: the unchanged parent compound, a cysteine adducts, and an unstable, chemically uncharacterized metabolite [9].
LDs display a steep dose–response curve. Below a certain plasma concentration (threshold), the natriuretic and diuretic effects are minimal; however, above this threshold, the response increases rapidly. At higher concentrations, a plateau is reached, wherein further increases in plasma concentration do not correspond to an enhanced effect. This dose–response relationship underpins the recommendation to double the dose if there is no response to initial diuretic administration [7, 8, 13].
In critically ill patients, however, the threshold for reaching the plateau is difficult to define owing to altered extracellular volume expansion and pathologic organ functions, which affects the pharmacokinetics and pharmacodynamics of the diuretics. For instance, the area under the curve for a specific intravenous dose of furosemide and a theoretical equipotent oral dose may be comparable; nevertheless, the time to surpass the natriuretic threshold can vary owing to pathological conditions affecting this threshold. This variability explains why intravenous doses of LDs that achieve higher peak levels may remain effective when equipotent oral doses lose efficacy [8, 13].
Indeed, the natriuretic threshold, defined as the luminal concentration necessary to elicit a significant diuretic response, depends not only on pharmacokinetics parameters but also on the actual patient’s clinical conditions or comorbidities. In critically ill patients, fluid accumulation, organ edema, and impaired cardiac or renal function notably impact LD efficacy. Intestinal edema, as occurs also in heart failure or nephrotic syndrome, but simply owing to fluid accumulation, can impair the enteral absorption of LDs, necessitating intravenous administration to avoid diuretic resistance. Furthermore, as in heart failure, an impaired cardiac index leads to altered renal perfusion, increased intraparenchymal pressure, and neurohormonal activation. These conditions result in diuretic resistance (DR), and the necessity of higher doses or combination therapies to overcome diminished responsiveness.
In acute or chronic kidney disease, decreased glomerular filtration rate (GFR) and competition with endogenous anions for OAT-mediated secretion elevate the threshold dose needed for efficacy. About 15–20% of the furosemide dose is delivered into the tubular fluid in patients with stage 5 CKD. This diminished tubular secretion is due to the elevated level of endogenous organic anions that interfere with furosemide secretion via organic acid transporters in the proximal tubule [14].
Hypoalbuminemia reduces the plasma concentration of loop diuretics available for secretion into the renal tubules. Additionally, the substantial protein loss in urine (proteinuria) might lead to loop diuretics binding to albumin within the tubular fluid, potentially reducing their availability to interact with the Na+-K+-2Cl− cotransporter in the thick ascending limb [15].
Collectively, these conditions underscore the importance of understanding the complex interplay between LD pharmacology and pathophysiological states to optimize therapeutic outcomes.
LDs can cause hearing loss in human at very high doses > 1 g or infusion rate > 250 mg/h; this risk is greater in patients with GFR < 20 ml/min/1.73m2 or concurrent administration of other ototoxic drugs (e.g., gentamicin and cisplatin) [7]. Furosemide ototoxicity involves NKCC-2 cotransporters in the inner ear stria vascularis encoded by the gene SLC12A2 and seems to be more linked to speed to infusion and the peak levels than the total dose. Deafness usually occurs 10–20 min after intravenous administration and is usually reversible and of short duration (1–24 h) [9].
LDs led to distinct pathological alterations in the cochlea, notably the development of oedematous spaces within the stria vascularis epithelium. This process results in a swift decline of the endolymphatic potential and ultimately causes the loss of cochlear microphonic potential, summating potential, and compound action potential. These diuretics also affect strial adenylate cyclase and Na+/K+-ATPase while inhibiting the Na+-K+-2Cl− cotransporter found in the stria vascularis. However, recent findings suggest that one of the earliest observable effects in vivo is a reduction of blood flow to the vessels supplying the cochlear lateral wall. Although EA does not result in damage to the stria vascularis under laboratory conditions, the changes noted in Na+/K+-ATPase and the Na+-K+-2Cl− cotransporter in living organisms may be secondary effects arising from ischemia and anoxia in the stria. Additionally, recent studies have revealed the presence of renin in pericytes surrounding the stria arterioles, indicating that diuretics may cause localized vasoconstriction through the secretion of renin and subsequent formation of angiotensin. The tight junctions of the blood–cochlea barrier serve to protect the cochlea from toxic molecules and pathogens. However, when diuretics provoke a temporary ischemic state, this barrier can become compromised, allowing harmful substances or pathogens to penetrate the cochlea [16]. These factors may account for instances of permanent hearing loss following the administration of EA, contributing to its diminished clinical application [9]. Angiotensin-converting enzyme (ACE) inhibitors may provide a protective effect against cochlear ischemia and damage caused by ototoxic drugs, such as cisplatin [17]. Blocking this system could reduce ischemic damage and oxidative stress in the cochlea, potentially lowering the risk of ototoxicity. However, their protective effect in the context of furosemide ototoxicity has not been extensively explored. Oxygen inhalation, the coadministration of triamterene (a potassium-sparing diuretic), iodinated benzoic acid derivatives such as diatrizoate and probenecid, and organic acids such as sodium salicylate and penicillin G have been demonstrated to attenuate the ototoxicity from diuretics. Despite these potential interventions, pharmacological treatments are generally not of major clinical significance, as the ototoxicity caused by diuretics is typically short-lived [18].
LDs can cause hyperuricemia and hyperglycemia and can increase the level of low-density lipoprotein cholesterol and triglycerides while decreasing high-density lipoprotein cholesterol. All these actions are reversible upon cessation of administration. Allergic interstitial nephritis may occur with chronic use of sulfa-containing LDs [9].
Furosemide and bumetadine contain a sulfonamide moiety, whereas torsemide is a sulfonyl urea. In patients who are suspected to be allergic to sulfa, EA should be used because it does not contain sulfa [9].
3.3 Diuretic ResistanceDiuretic resistance occurs when a patient with fluid overload becomes refractory to conventional diuretic therapy, regardless of whether the patient is naïve to diuretics or has been previously treated. The factors contributing to diuretic resistance can be categorized into three main groups: (1) pharmacokinetic properties of the drugs, (2) coadministration of other medications that inhibit the action of loop diuretics, and (3) coexistence of organ failure, particularly acute or chronic renal failure.
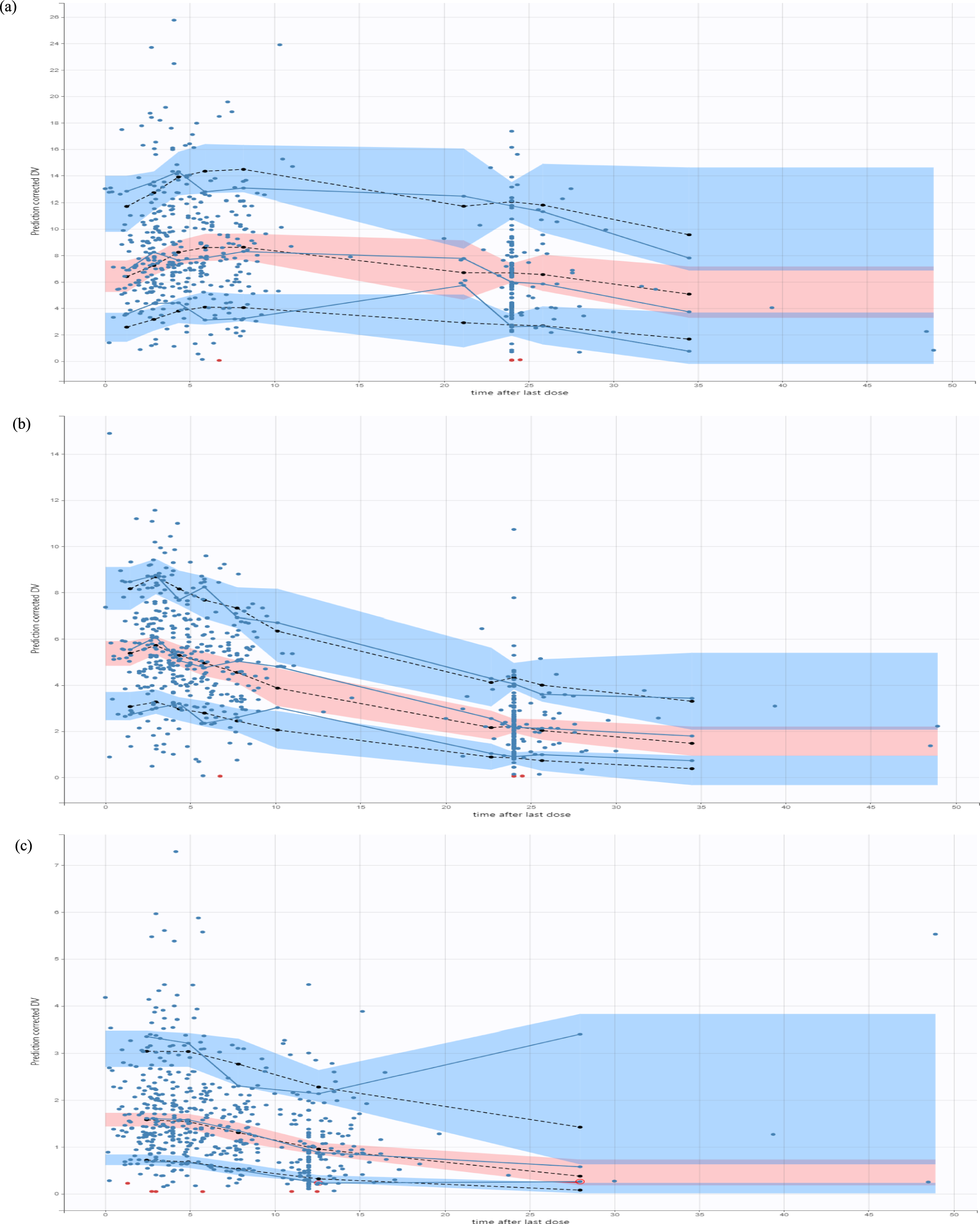
Owing to their short half-life, initial natriuresis typically diminishes within the first 3–6 h following administration, resulting in an interval (16–21 h) in which the diuretic effect is essentially absent, but sufficient for the kidneys to compensate the acute electrolyte and water losses that occur after diuretic administration [8, 23]. At steady state, postdiuretic Na+ retention may be observed, characterized by excretion rates falling below baseline levels once the effects of the previously administered diuretic have waned. This condition persists until a subsequent diuretic dose is administered. Following any therapeutically active dose, natriuresis tends to decrease as extracellular fluid volume diminishes, a phenomenon commonly referred to as “braking,” which is particularly noted in patients undergoing chronic diuretic treatment. The phenomenon underscores the presence of both functional and structural adaptive processes in critically ill patients [8, 23]. Specifically, there is an upregulation of proteins involved in renal Na+ absorption [23]. Consequently, the increased release of Na+ into the distal tubular segments, which occurs owing to the inhibition of NKCC-2 in the ascending limb of the loop of Henle, subsequently stimulates the activity of Na+/Cl− channels. This process is also linked to an elevated number of Na+/K+-ATPase pumps in the basolateral membrane of tubular cells. These structural modifications significantly impair the effectiveness of LDs, particularly in patients with chronic diuretic therapy [8, 23]. In summary, the braking phenomenon represents a remodeling of the distal nephron, characterized by hypertrophy and hyperplasia resulting from increased Na+ release, elevated levels of angiotensin II and aldosterone, and alterations in K+ balance. The consequences of this remodeling include an enhanced transport capacity of the distal tubules, enabling them to compete more effectively with the ascending loop of Henle [8, 23]. Consequently, as more Na+ is extruded from the loop, a greater amount is reabsorbed distally, leading to a decrease in net natriuresis [8, 23]. Functional compensatory response is postdiuretic sodium retention, where the kidneys avidly retain sodium after the diuretic effect of a LD has waned, counteracting the initial sodium loss. This can be triggered by the activation of the renin–angiotensin–aldosterone system (RAAS) and the sympathetic nervous system in response to the diuretic-induced volume changes. Increased proximal tubular sodium reabsorption has also been observed as a compensatory mechanism that can limit the overall efficacy of loop diuretics. Chloride depletion, a potential consequence of LD therapy, can contribute to diuretic braking by stimulating sodium reabsorption in the distal nephron. Furthermore, the increased delivery of sodium to the macula densa as a result of loop diuretic action can trigger tubuloglomerular feedback, leading to a reduction in GFR and renal blood flow, which may subsequently limit the diuretic efficacy. These compensatory renal mechanisms highlight the dynamic interplay between loop diuretics and the kidney’s intrinsic regulatory systems, often contributing to diuretic resistance and necessitating adjustments in treatment strategies (Fig. 2). To mitigate this adaptive response and restore diuretic efficacy, it is beneficial to add a second diuretic from a different class, such as a thiazide diuretic.
Fig. 2
Mechanisms of diuretic resistance. The kidney employs several compensatory mechanisms that can influence the response to loop diuretics, particularly with chronic use. Furthermore, coadministration of other medications, and coexistence of organ failure, particularly acute or chronic renal failure, can affect the loop diuretics’ effects
OAT type 1 and 3 can be inhibited by coadministered drugs, particularly nonsteroidal antiinflammatory drugs (NSAIDs). The mechanism by which LD inhibit Na+ reabsorption involves their action at the level of the macula densa. This inhibition stimulates the secretion of renin and promotes the production of prostaglandins through cyclooxygenase 2. Prostaglandin E2, in turn, acts on the renal tubules, enhancing natriuresis and inhibiting Na+ reabsorption along the ascending limb of the loop of Henle and within the collecting duct. However, NSAIDs block this prostaglandin-mediated action [7, 8, 13, 23]. Reported half-maximal inhibitory concentration values indicate comparable affinities of OAT1 and OAT3 for diuretics, cephalosporins, and NSAIDs, whereas OAT2 shows generally lower affinity for these compounds [24, 25]. Furosemide and bendroflumethiazide have been shown to inhibit tracer uptake via mOat1 and mOat3 in a concentration-dependent manner, consistent with their behavior as substrates of these transporters [25]. Consequently, drug–drug interactions at OAT1 and OAT3—particularly with coadministered β-lactam penicillins or thiazide diuretics—may impair renal drug clearance and increase the risk of adverse effects. Moreover, furosemide-induced alterations in tubular ion transport may further influence the reabsorption of other positively charged compounds, such as certain antibiotics, through effects on the luminal electrochemical gradient.
LDs also inhibit NKCC-1 channels, which are expressed in vascular smooth muscle cells. This inhibition contributes to afferent arteriolar vasodilation, helping to maintain glomerular filtration rate (GFR) even in the context of reduced extracellular volume. Importantly, this compensatory adaptation relies on the production of prostaglandins, an effect that can be compromised using NSAIDs [13, 23].
Several studies have reported a high incidence of DR, ranging from 50% to 70%, in patients with renal syndromes [3]. OATs in the ascending limb of the loop of Henle and the proximal convoluted tubule also mediate the reabsorption of uric acid and weak anions. Kidney failure and other conditions leading to persistent renal hypoperfusion can result in the progressive accumulation of organic anions, which compete with diuretics for active secretion and transport to their sites of action. Additionally, in the context of metabolic acidosis, the depolarization of the cell membrane may further reduce the activity of OATs [23].
3.4 How Can We Face Diuretic Resistance?From a practical perspective, two different issues arise in the management of critically ill patients regarding diuretic therapy: (1) determining the appropriate diuretic dose needed to surpass the efficacy threshold and (2) defining strategies to sustain diuresis in the absence of a response despite escalating doses. One previously mentioned approach involves combining LDs with a thiazide diuretic to mitigate the braking phenomenon. Alternatively, LDs can be administered alongside albumin, or they may be given as boluses rather than through continuous infusion. These strategies are inherently grounded in pharmacokinetic principles.
Diuretic resistance (DR) is associated with an elevated risk of mortality [26], particularly in patients in the ICU with severe heart failure or fluid overload. The assessment of DR relies on a combination of the clinician’s subjective evaluation of inadequate drug efficacy and quantitative indicators from the patient, such as urine output. However, there is presently no universally accepted definition of DR [3] that can be consistently applied in clinical practice or utilized in the design of clinical studies.
In a retrospective study that used the publicly available deidentified Medical Information Mart for Intensive Care III (MIMIC-III) database, Coté et al. considered adult patients admitted to ICU for more than 24 h and treated with a dose of furosemide greater than 1 mg/kg/24 h. They explored the effectiveness of the following interventions: mode of furosemide administration (continuous infusion versus intermittent), incremental dosing of diuretics, use of albumin, or addition of a second class of diuretics. The primary endpoint of the study was diuresis within the first 24 h, while secondary efficacy endpoints included fluid balance over 24 h and weight change at 24 and 48 h. Safety endpoints encompassed electrolyte abnormalities and acid–base balance. The results showed that doses of furosemide greater than 2, 3, or 4 mg/kg/die resulted in a progressive increase in urine output over 24 h. Additionally, continuous infusion was associated with greater urine output over 24 h, improved fluid balance, and greater weight loss within the same period [27]. The addition of a second diuretic—such as thiazides, mineralocorticoid antagonists, or carbonic anhydrase inhibitors—resulted in a modest increase in 24-h urine output and varied according to the class of diuretic administered, with thiazide diuretics demonstrating a more pronounced urine output. Additionally, the combination of furosemide with albumin was associated with a positive fluid balance, a reduction in diuresis over the 24-h period, and no significant change in body weight [27].
Continuous infusion of furosemide represents a strategic approach to mitigate the effects of the drug’s pharmacokinetics. There are several compelling reasons to consider continuous infusion of furosemide: it minimizes fluctuations in serum concentration, addresses the limitations associated with the drug’s short half-life, facilitates more predictable and consistent diuresis, mitigates the risk of developing diuretic resistance by eliminating the postdiuretic rebound effect that may activate compensatory renal mechanisms, and ultimately enhances hemodynamic stability in the patient.
In a meta-analysis focusing on cardiac patients, the primary efficacy endpoint of diuretic therapy was the change in body weight, despite this parameter is typically regarded as a secondary endpoint in clinical practice. Notably, continuous infusion of furosemide was shown to be more effective than bolus administration [28].
Ng et al., in a meta-analysis involving patients in the ICU, assessed not only the different methods of furosemide administration (continuous infusion versus bolus administration) but also an adaptive dosing strategy based on the observed diuretic response (increasing or decreasing the dose as necessary). They found that the dose-adjustment strategy was superior in enhancing diuresis compared with the mere comparison of continuous infusion versus bolus administration, especially in hemodynamically unstable patients. Furthermore, subgroup analyses revealed that this targeted therapeutic approach was associated with a reduction in ICU length of stay compared with patients who did not receive this strategy [29].
It is crucial to highlight that all studies included in these meta-analyses identified ICU and hospital mortality as primary endpoints related to the administration of furosemide, whether given as a continuous infusion or in bolus form. Consequently, the sample size calculations in these studies were based on these endpoints, rather than on measures of diuretic efficacy. Furthermore, the target populations exhibited considerable heterogeneity, as did the protocols for furosemide administration.
Another explored strategy to mitigate DR involves the administration of furosemide in combination with albumin. LDs, particularly furosemide, are bound by approximately 95% to plasma proteins, with albumin as principal transporter. In cases of hypoalbuminemia, the distribution volume of furosemide increases, resulting in a reduced amount of albumin-bound furosemide available for the proximal tubules. As a result, in patients with severe hypoalbuminemia, the efficacy of the diuretic may be compromised. Elevating albumin levels prior to or concurrently with furosemide administration may enhance its therapeutic effect.
Studies have indicated that the infusion of albumin alone can lead to increased diuresis, likely owing to an elevation in intravascular volume, improving hemodynamic parameters [30] and consequent increase in the estimated GFR (eGFR) and the effective renal plasma flow [31, 32]. A meta-analysis encompassing only randomized controlled trials, albeit heterogeneous in terms of target populations, compared the combined administration of furosemide and albumin to albumin alone. This analysis revealed that coadministration resul
Comments (0)