
Remember me
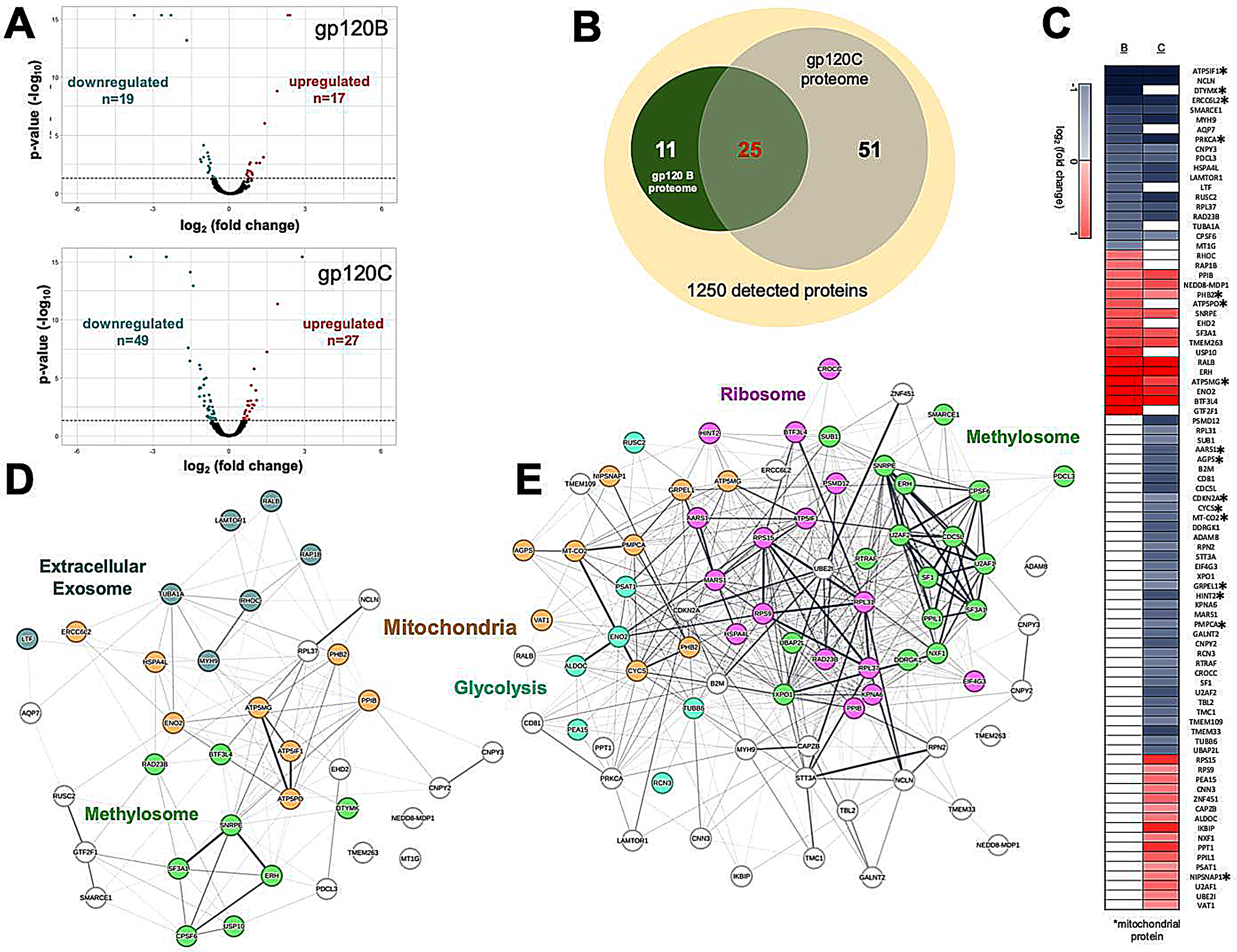

The chemical composition of C. subedentata extract was previously characterized in studies by Cabezas et al. and Castillo et al. [24, 25]. The principal compounds identified and isolated from the extract include galantamine (24.77%), maritidine (21.45%), tazettine (12.45%), galantindol (8.45%), haemanthamine (5.45%), ismine (4.27%), narwedine (3.79%), lycorine (2.73%), deoxytazettine (2.32%), and kirkine (0.40%). Additional minor metabolites identified were trisphaeridine, 11,12-dehydroanhydrolycorine, homolycorine, and anhydrolycorine. The concentrations of C. subedentata extract used in the test were consistent with those reported in early studies [25].
Caliphruria subedenta Extract Offers Protection Against Neurotoxicity Induced by Aβ (1–42) Peptide and Okadaic AcidAβ (1–42) peptide and OA are well-established neurotoxic agents used in various in vitro and in vivo models. Neurotoxicity induced by these compounds is widely accepted as a model for mimicking neuronal death associated with AD. Aβ (1–42) peptide, a biochemical hallmark of neurodegeneration, is believed to result from aberrant processing of amyloid precursor protein (APP). In contrast, OA, a toxin produced by marine algae, inhibits protein phosphatases, leading to increased tau phosphorylation and neuronal death. To evaluate whether pretreatments or co-treatments with C. subedenta extract provides significant neuroprotection against these neurotoxic stimuli, SH-SY5Y cells were treated for 24 h with C. subedenta extract at concentrations of 6.25, 12.5, and 25 µg/mL, followed by 24 h exposure to Aβ (1–42) (10 µM) or OA (30 nM). Under our experimental conditions, pre-treatments with C subedentata extract at 6.25 and 12.5 µg/mL significantly increased the cell survival rate in response to Aβ (1–42) and OA-induced neurotoxic (Fig. 1).
Fig. 1
C. subedentata extract offers protection against Aβ(1–42) and OA-induced neurotoxicity in the human neuroblastoma cell line SH-SY5Y. SH-SY5Y cells were treated for 24 h with increasing concentration (6.25, 12.5 and 25 µg/mL) of C. subedentata extract, then, the cells were incubated with a Aβ(1–42) (10 µM) and b OA (30 nM) to induce neurotoxicity for 24 h. Data are means ± standard deviation (S.D) calculated for three independent experiments each one performed in triplicate. **, p < 0.01 and ***, p < 0.001 in comparison with injured cells by Aβ (1–42) and OA. Co, cells not treated
Under different experimental conditions, SH-SY5Y cells were treated simultaneously with C. subedenta extract and neurotoxic stimulus (Aβ (1–42) at 10 µM or OA at30 nM. As shown in Fig. 2, both Aβ (1–42) and OA significantly decreased cell survival (p < 0.001). However, co-treatment with the extract at concentrations of 6.25, 12.5 and 25 µg/mL led to significant increases in cell survival compared to the control.
Fig. 2
Simultaneous treatments with C. subedentata extract and neurotoxic stimuli decrease the neurotoxicity in SH-SY5Y cells. SH-SY5Y cells were incubated during 48 h with increasing concentration (6.25, 12.5 and 25 µg/mL) of C. subedentata plus Aβ (1–42) (10 µM) (a) or OA (30 nM) (b). Data are means ± standard deviation (S.D) of triplicate of three different experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 in comparison with injured cells by Aβ (1–42) and OA. Co, cells not treated
These results demonstrate that both Aβ (1–42) and OA significantly reduce the proliferative capacity of SH-SY5Y cells. The pathological hallmarks of AD include the deposition of Aβ (1–42) and the accumulation of hyperphosphorylated tau protein [42]. In the context of Aβ (1–42)-induced cell death, Aβ is recognized as the primary constituent of senile plaques in AD and plays a pivotal role in its pathogenesis. Increasing evidence indicates that Aβ induces oxidative damage to proteins, disrupts calcium homeostasis by penetrating the mitochondrial matrix, and progressively accumulates to induce mitochondrial stress. This stress interferes with enzymatic activity and impacting neuronal bioenergetics [43, 44]. Consequently, these process lead to ATP depletion, which is critical for mitotic functions and cell death pathways. Additionally, Aβ exacerbates genotoxic damage in SH-SY5Y cells [25, 45, 46].
OA serves as a robust model for mimicking tau hyperphosphorylation in AD. This toxin inhibits phosphatases, displaying the highest affinity for PP2A followed by PP1 and PP2B. Its effects include inducing hyperphosphorylation in both in vivo and in vitro [47, 48]. Tau proteins isolated from AD brains exhibit over 40 phosphorylation sites with enzymes such as glycogen synthase kinase (GSK-3β) and protein phosphatase 2A (PP2A) playing critical roles in tau phosphorylation and dephosphorylation respectively [49]. In SH‐SY5Y cells, phosphorylation of GSK-3β at Ser9 reduces its activity, while phosphorylation at Tyr216 increases it [50]. The association between tau hyperphosphorylation and neuronal death through PP2A inhibition and GSK3 activation by OA has been documented in cortical and hippocampal neuron cultures, as well as SH-SY5Y cells [51,52,53]. Conversely, GSK3 inhibitors have been shown to reduce tau hyperphosphorylation. Research into the mechanisms of AChEIs has expanded beyond AChE inhibition to include their ability to mitigate cell damage by decreasing Aβ deposition [54]. Regarding the neuroprotective mechanisms of C. subedentata extract during pre and co-treatment against Aβ (1–42)-induced neurotoxicity, it has been reported that post-treatments with the extract reduces neuronal death by inhibiting AChE, decreasing genotoxic damage, and regulating the mitochondrial morphology [25]. Since neuronal homeostasis depends closely on mitochondria motility and function-crucial for differentiation processes [55, 56], it is plausible that C. subedentata alkaloids influence key energetic pathways involved in differentiation. Additionally, treatments with C. subedentata have been shown to promote neural differentiation effectively, likely through epigenetic regulation involving, with histone deacetylases [57]. Overall, our findings demonstrate that C. subedentata extract exerts protective effects during pre-treatment, co-treatment, and post-treatment [25] against neurotoxic stimul. However, these cannot be attributed solely to alkaloids, as non-alkaloid compounds may also contribute to their neuroprotective action. These results underscore the importance of exploring strategies aimed at not only treating degenerative pathologies but also on preventing them.
Molecular Docking and Therapeutic ViabilityNowadays, due to the impact of AD on patients, caregivers and society, there is a growing need to find treatment or cure, and this way, reducing worldwide the effects of pathology. Because prevalence of AD is strongly associated with increasing age, it is expected that this dementing disorder will pose huge challenges to public health and elderly care systems worldwide [58]. To date, drug discovery efforts for AD have achieved limited success, primarily in the development of symptomatic treatments. These include AChEIs such as donepezil, rivastigmine, galantamine and memantine, an N-methyl-D-aspartate receptor (NMDA) antagonist. While, these drugs offer palliative benefits, they do not alter the progression of the disease. This failure in developing effective treatments can be attributed to the complex, multifactorial nature of AD pathogenesis and the absence of adequate and validated biomarkers, which significantly hinder the development of targeted therapies [3].
In this study, molecular docking was employed to evaluate the binding affinity between four targets (AChE, BuChE, NMDA and GSK3) and a selection of alkaloids ismine, kirkine, maritidine and tazettine, anhydrolycorine, homolycorine narwedine and galanthindole. Positive controls used for comparison included galantamine, tacrine, memantine, 6LQ and ATP. All tested alkaloids were capable of forming stable complexes. The docking scores for each complex are summarized in Table 1. Among the alkaloids anhydrolicorine, maritidine and galanthindole exhibited the highest average docking scores (60.08, 59.74, and 58.69 respectively), surpassing the score of galantamine (57.97). For the BuChE target, all alkaloids, except for ismine, demonstrated mean docking score values exceeding that of the positive control tacrine (52.50). In the case of the NMDA receptor, all ligands achieved higher mean scores values compared to memantine (40.67). Regarding the GSK3 target, kirkine exhibited the highest docking score among the alkaloids (57.56), however, this value did not surpass the scores observed for the positive controls 6LQ (83.77) and ATP (62.35).
Table 1 GOLD Mean scores. Molecular docking scores of ligands with target receptorsTo evaluate the stability of the complexs, we focused on the ligands with the highest docking scores: anhydrolycorine/AChE = 60.0; galanthindole/BuChE (63.24); homolicorine/NMDA (57.87) and kirkine/ GSK3-β (57.56). The results of the protein–ligand interactions for the best pose of each ligand are summarized in Table 2 and illustrated in Fig. 3. The analysis revealed distinct interaction patterns across the complexes. The GS3K/Kirkine complex exhibited the highest number of hydrophobic interactions (six) and hydrogen bonds (three), highlighting its strong binding stability. In contrast, anhydrolycorine/AChE complex demonstrated the greatest number of pi-stacking interactions (three), emphasizing its unique model of binding. Notably, the NMDA/homolycorine complex showed a balanced distribution of hydrophobic interactions across its interaction profile. These findings suggest that the various complexes may have different mechanisms of action, depending on the specific interactions they establish with their ligands.
Table 2 Protein–ligand interactions of the best pose for each ligand and patterns of interactions between the complexes and their ligandsFig. 3
Top: Molecular interaction between ligands and targets
By identifying the specific interactions between a protein target and their ligands, we can gain valuable insights into the underlying mechanisms of binding and the potential functional effects of these interactions. This knowledge serves as a foundation for designing and optimizing ligands or drugs with enhanced binding properties, ultimately leading to therapeutic outcomes. The molecular interactions identified in the analyzed complexes (Table 2 and Fig. 3), including hydrophobic interactions, hydrogen bonds, π-stacking, and salt bridges, each play distinct roles in stabilizing ligand binding and enhancing specificity. Hydrophobic interactions are predominant across all complexes, acting as the primary mechanism for anchoring ligands within the hydrophobic pockets of the target proteins. Hydrogen bonds, which are critical contributors to binding specificity and stability, were particularly prominent in the NMDA/Homolycorine and GSK3β/Kirkine complexes, highlighting their essential role in molecular recognition. π-Stacking interactions, observed in complexes featuring aromatic residues in the binding site, such as AChE and BuChE, further enhanced stabilization through aromatic-aromatic interactions. Salt bridges identified in the NMDA/Homolycorine complex provided electrostatic complementarity, thereby strengthening the ligand–protein interaction. This understanding of interaction profiles provides a solid foundation for designing ligands tailored to exploit these molecular features. For instance, insights into the predominant hydrophobic interactions or hydrogen bonds within a specific complex facilitate the rational design of ligands optimized for these interactions, potentially leading to more effective binding and superior therapeutic outcomes. Consequently, the results presented in Table 2 and Fig. 3 serve as valuable guidance for the development of novel ligands or drugs that can selectively and effectively target each protein complex. For instance, knowledge of predominant hydrophobic interactions or hydrogen bonds in each complex enables the rational design of ligands optimized for these interactions, leading to more effective binding and potentially superior therapeutic outcomes. Therefore, the results presented in Table 2 and Fig. 3 are valuable for guiding the development of novel ligands or drugs that can selectively and effectively target each protein complex.
To evaluate the potential toxicity of the compounds under investigation, we used the ProTox-II server, and the results are presented in Table 3. This server predicts the likelihood of a compound causing carcinogenic, immunotoxic, mutagenic, and cytotoxic effects, while also estimating the LD50 value (in mg/kg body mass). The LD50 values generated by the server classify molecules based on their toxicity, ranging from Category I (indicating compounds that have the potential to be lethal) to Category VI (indicating compounds with a low risk of harm if ingested) [35]. Although kirkine showed the lowest LD50 value, indicating potential toxicity upon ingestion, it was classified as Category 3, suggesting moderate toxicity. In contrast, the other ligands were categorized as Category 4, indicating a level of toxicity that can be harmful if ingested.
Table 3 Prediction of toxicological properties analyzed by ProTox-IIDrug development presents a significant challenge for the pharmaceutical industry. It has been demonstrated that the primary factor contributing to the high failure rate of new chemical entities during drug development is not necessarily a lack of drug activity, but rather inadequate pharmacokinetic (PK) and pharmacodynamics (PD) properties. Gastrointestinal absorption and brain access are two critical pharmacokinetic processes that must be evaluated at various stages of drug discovery [59, 60]. Therefore, in addition to assessing the affinity and selectivity against molecular targets, we also evaluated absorption, distribution, metabolism, excretion and tolerable toxicity (ADMET). In the development of anti-Alzheimer drugs, a major limitation is overcoming blood brain barrier (BBB) permeation. The lack of BBB permeability prevents the active compound from reaching its target in the brain. The Brain Or Intestinal Estimated Permeation (BOILED-Egg) method is an accurate predictive model that computes the lipophilicity and polarity of small molecules [59]. The boiled-egg graphic in Fig. 4 presents the results of the assessment of human intestinal absorption (HIA) and BBB penetration. The yellow region indicates a high probability of permeation through the BBB, where seven molecules are located. Homolycorine and memantine are represented as red dots, indicating that they are P-gp non-substrates (P-gp-), whereas galantamine, kirkine, anhydrolycorine, galanthindole, tacrine, and ligand 6LQ are classified as P-gp substrates (P-gp +) (blue dot). It should be noted that ligand 6LQ is in the white region, indicating that it is passively absorbed by the gastrointestinal tract but still a P-gp+. One molecule, ATP, falls outside the range of the graphic, suggesting limited potential for gastrointestinal absorption and BBB penetration. The Log S value of -4.07 indicates that ATP has high water solubility and may be largely confined to extracellular space. The Log P o/w value of 0.35 suggests that ATP has low lipophilicity, which is consistent with the limited potential for HIA and BBB penetration. Considering this, it seems reasonable to conclude that the position of ATP out of range in the BOILED-Egg graphic is consistent with its limited potential for systemic exposure and its properties as a P-gp+ (Table 4).
Fig. 4
Boiled-egg graphic shows evaluation of passive gastrointestinal absorption (white area) and brain access (yellow area) of the alkaloids and positive-controls: #1Galantamine; #2 Kirkine; #3 anhydrolycorine; #4 homolycorine; #5 galanthindole; #6 tacrine; #7 memantine; #8 6LQ and #9 ATP
Table 4 Pharmacokinetic properties of the alkaloids tested by the SwissADME ServerLipinski's rule of five, as employed in SwissADME, elucidates the correlation between pharmacokinetic and physicochemical parameters of orally active compounds. This rule characterizes small molecule profiles based on four descriptors: molecular weight (MW) ≤ 500 Da, partition coefficient octanol–water (MLOGP) ≤ 4.15, number of hydrogen bond acceptors (N or O) ≤ 10, and number of hydrogen bond donors (NH or OH) ≤ 5 [59]. (Table 4).
Galantamine adheres to Lipinski's rule of five, exhibiting moderate solubility (2.64 mg/L) and moderate lipophilicity (-2.93 Log P O/W). Similarly, Kirkine also conforms to Lipinski's rule, showing moderate solubility (2.42 mg/L) and moderate lipophilicity (-2.25 Log P O/W). Anhydrolycorine also complies with the rule demonstrating moderate solubility (2.63 mg/L) and high lipophilicity (-3.81 Log P O/W). Homolycorine adheres to Lipinski's rule, with moderate solubility (2.98 mg/L) and moderate lipophilicity (-3.09 Log P O/W). Galanthindole, tacrine, and memantine also conform to Lipinski's rule, with moderate solubility values (2.77 mg/L, 2.09 mg/L, and 2.51 mg/L, respectively) and moderate lipophilicity (-3.59 Log P O/W, -3.27 Log P O/W, and -3.02 Log P O/W, respectively). In contrast, 6LQ and ATP do not fully comply with Lipinski's rule. 6LQ exhibits low solubility (3.29 mg/L) and very high lipophilicity (-4.07 Log P O/W), while ATP demonstrates high solubility (0.93 mg/L) but low lipophilicity (0.35 Log P O/W). Consequently, according to Lipinski's rule, galantamine, kirkine, anhydrolycorine, hololycorine, galanthindole, tacrine, and memantine are considered toconform, while 6LQ and ATP do not. Based on these results, most of the ligands in Table 4 (galantamine, kirkine, anhydrolycorine, hololycorine, galanthindole, tacrine, and memantine) comply with Lipinski's rule. This suggests a higher probability of oral bioavailability for these ligands. The ligands generally exhibit moderate solubility in water, ranging from 2.09 mg/L to 3.29 mg/L. However, it is important to note that 6LQ demonstrates relatively low solubility compared to the other ligands. The lipophilicity of the ligands varies, with 6LQ and galanthindole demonstrating high lipophilicity, while ATP exhibits low lipophilicity. Lipophilicity influences a drug's ability to permeate cell membranes and distribute throughout the body. The molecular weights of the ligands range from 179.30 g/mol to 535.66 g/mol. Larger molecules, such as 6LQ, may encounter challenges in absorption and distribution. In conclusion, ligands that adhere to Lipinski's rule possess moderate solubility, and moderate lipophilicity are more promising candidates for further drug development, with a higher probability of favorable pharmacokinetics properties.
Molecular DynamicsDynamics simulations were conducted to assess the molecular behavior of the complexes over time. To evaluate the stability of each system, we analyzed the RMSD, which measures the average distance between the atoms of each protein over the simulation frames. Lower RMSD values indicate that the structure remains close to its initial conformation, while higher values suggest significant deviations. A stable system typically shows a plateau in RMSD values, indicating that the complex has reached equilibrium, and its structure is no longer undergoing large fluctuations. Figure 5 illustrates the RMSD of cholinesterases (AChE and BuChE), NMDA and GSK3. The trajectories of these enzymes interacting with the ligands stabilize over the course of the simulation. The complexes involving AChE, BuChE and GSK3 reached stability around 10 ns, while the NMDA system took a longer time to stabilize, with equilibrium achieved around 50 ns. The complexes with cholinesterases and GSK3 exhibited RMSD values close to 0.18 nm (Figs. 5a–c). In contrast, for the NMDA complexes, the homolycorine ligand initially showed high RMSD values, but these values stabilized near 0.25 nm after 50 ns, resembling the behavior of the NMDA-Memantine complex (Fig. 5d).
Fig. 5
Backbone RMSD as a function time: a trajectories of compounds interacting with AChE; b with BuChE; c with GSK3 and d with NMDA
To assess the amino acid residues contributing to fluctuations in trajectories, we analyzed the RMSF of each complex (Fig. 6). RMSF quantifies the flexibility of residues by measuring their average deviation from a reference position over time, highlighting regions of the protein that exhibit greater or lesser mobility. High RMSF values indicate more flexible regions, while low values correspond to more rigid and stable regions. The AChE complexes exhibited similar fluctuation patterns, with notable peaks for residues TYR77 and LEU221 being higher when AChE was associated with the anhydrolycorine ligand (Fig. 6a). For BuChE the complexes with both ligands showed comparable fluctuations throughout the simulation (Fig. 6b). Regarding the GSK3 complexes, the 6LQ ligand induced smaller fluctuations, particularly in the MET250-ILE262 loop (Fig. 6c). In contrast, the NMDA complexes demonstrated higher RMSF values across most amino acid residues with the NMDA-homolycorine complex exhibiting the greatest fluctuation (Fig. 6d).
Fig. 6
RMSF as function amino acid residues: a trajectories of compounds interacting with AChE; b with BuChE; c with GSK3 and d with NMDA
When analyzing the RMSD of the ligands, we observed that most ligands exhibited RMSD values greater than 0.2 nm across the analyzed enzymes (Fig. 7). The only exception was galantamine interacting with AChE, which displayed a trajectory with peaks below this threshold, stabilizing at approximately 0.08 nm (Fig. 7a). However, the higher RMSD values observed for the other ligands suggest that their positions varied throughout the simulation. In the case of the BuChE complexes, although the trajectories shown peaks above the threshold, the tacrine and galanthindole ligands tend to stabilize at approximately 0.5 and 0.6 nm, respectively (Fig. 7b). Likewise, kirkine, ATP, and 6LQ tend to stabilize around 0.5, 0.7 and 0.4 nm, respectively (Fig. 7c). On the other hand, the trajectories of the AChE-anhydrolycorine complex (Fig. 7a) and of the NMDA complexes (Fig. 7d) exhibited significant fluctuation throughout the simulation, suggesting that these ligands may be subject to dissociation events.
Fig. 7
Ligand RMSD as a function time: a trajectories of compounds interacting with AChE; b witch BuChE; c with GSK3; and d with NMDA
Trajectory frames indicated that although the AChE-anhydrolycorine complex and the systems formed by BuChE and GSK3 exhibited RMSD peaks above the 0.2 nm threshold, these ligands remained tightly bound to their respective binding sites. Specifically, galantindole and kirkine, natural products associated with BuChE and GSK3 respectively, showed only minor conformational variations over time. However, they remained anchored within the active sites of these enzymes, stabilized by hydrogen bonding and π-stacking interactions, and surrounded by hydrophobic residues. Figure 8 illustrates representative frames of these ligands. Figure 8 illustrates representative frames of these ligands.
Fig. 8
Trajectories of frames of Galanthindole bound with BChE and Kirkine linked with GSK3. Proteins are represented by the color gray in the cartoon format and the ligands have carbon atoms customized in green in the sticks format
In contrast, the trajectories of anhydrolycorine associated with AChE, along with the ligands interacting with the NMDA receptor, showed susceptibility to dissociation events. The dissociation of these ligands occurs at specific time for each complex (Fig. 9. Specifically, anhydrolycorine began to dissociation from the AChE active site at approximately 29 ns, while homolycorine and memantine initiate dissociation from the NMDA site at around 5 ns and 4 ns, respectively. Given that homolycorine and memantine dissociatefrom NMDA and anhydrolycorine leavs AChE site, we proceed to analyze the radius of gyration (Rg), solvent-accessible surface area (SASA), and free energy calculations (MM/PBSA) of those ligands that remained docked to their enzyme.
Fig. 9
Trajectories of frames of anhydrolycorine bound with AChE and homolycorine and memantine linked with NMDA. Proteins are represented by the color gray in the cartoon format and the ligands have carbon atoms customized in green in the sticks format
To assess the overall compaction of the proteins that remained interacting with their ligands, the Rg was calculated (Fig. 10). Rg provides information about protein compaction, indicating whether the protein reached conformational equilibrium during the simulations. Figure 10a shows that the trajectory of the AChE-galantamine system equilibrates at approximately 2.32 nm, while Fig. 10b illustrate that the BuChE complexes interacting with galanthindole and tacrine reach Rg values around to 2.34 nm, remaining stable throughout the simulations. This suggests that these protein structures do not undergo significant fluctuation in compaction. Although the GSK3-β complexes exhibited subtle Rg variations (Fig. 10c), the trajectory showed a consistent compaction pattern for the tested ligands. The interaction with kirkine, ATP and the 6LQ presents tenuous compression and decompression movements; however, the trajectories tend to reach equilibrium close to 2.18 nm.
Figure10
Rg as a function time: a trajectories of compounds interacting with AChE; b with BuChE and c with GSK3
SASA analysis allows describing the molecular surface of proteins that are accessible to the solvent. The systematic increase in SASA suggests destabilization of the protein, which may expose its hydrophobic regions to the solvent. In general, the SASA values of the AChE, BuChE and GSK3 enzymes stabilized throughout the simulation (Fig. 11). The trajectories of the AChE/anhydrochorine complex stabilized at approximately 219 nm2 (Fig. 11a), while the complexes involving the BuChE enzyme demonstrated SASA values close to 226 nm2 (Fig. 11b). The GSK3 trajectories demonstrated subtle differences from SASA, where lower values were found when the enzyme interacts with the 6LQ (~ 183 nm2) and higher values when it interacts with ATP and kirkine, both with approximately 187 nm2 (Fig. 11c).
Fig. 11
Protein solvent-accessible surface area as a function time: a trajectories of compounds interacting with AChE; b with BuChE and c with GSK3
Through the MM/PBSA analysis, we predicted the binding energies for the ligands that continued to interact with their targets respectively (Table 5). Galantamine was the only compound that remained bound to AChE, showing a binding energy of − 346.30 ± 20.02 kJ.mol−1. This strong interaction highlights the potential of galantamine as a lead compound in AChE inhibition, suggesting that its binding profile could serve as a reference for the design of analogues with greater efficacy and selectivity. For the BuChE complexes, the galanthindole ligand exhibited a greater potential to compete for the enzyme´s active site, as it demonstrated a lower binding energy (− 78.15 ± 10.52 kJ.mol−1) compared to tacrine (− 68.56 ± 8.28 kJ.mol−1). These results highlight the importance of electrostatic interactions in stabilizing galanthindole within the BuChE active site, providing a promising framework for optimizing inhibitors targeting BuChE-related diseases such as AD. Structural modifications that improve these interactions could further enhance binding affinity and specificity. Regarding the GSK3 complexes, kirkine displayed higher binding energy values (− 227.90 ± 10.04 kcal.mol−1) compared to ATP and the 6LQ, which had scores of − 881.96 ± 26.27 and − 351.19 ± 13.12 kJ.mol−1, respectively. Although Kirkine binding energy is less favorable than ATP, its distinct interaction profile may inspire the design of GSK3 inhibitors capable of selectively modulating its activity, potentially minimizing off-target effects. These findings suggest that MM/PBSA-derived insights into binding energy, together with structural characterization of key interactions, can guide rational drug design by identifying functional groups or moieties essential for binding.
Table 5 Residual decomposition and Binding energy (given in kJ mol−1) in the protein–ligand interactionTaken together, these in vitro and in silico results provide evidence for the neuroprotective effects exerted by plants. Among the Amaryllidaceae family, galantamine-type alkaloids are widely recognized for their potential utility in treatment AD. In recent years, the multifactorial nature of AD has driven an active search for multi-target drugs with two or more selected biological activities, as they could represent a significant pharmacological advancement in managing this complex disease. In this context, drug combinations or compounds that may act at different levels of the neurotoxic cascade may offer new hope for treating neurodegenerative diseases like AD [64,65,66]. The findings from our study regarding the neuroprotective effects of C. subedentata extract align with the current trends in drug discovery for AD, which increasingly emphasize a multi-target approach over the traditional single-target strategies. This approach recognizes the multifactorial nature of AD, where a combination of neurodegenerative processes, such as cholinergic deficits, am
Comments (0)