
Remember me

This study was a three-phase, one-way crossover clinical trial in patients using trifluridine/tipiracil, performed at the Erasmus MC Cancer Institute, Rotterdam, the Netherlands [14]. The study protocol was written in conformity with the Declaration of Helsinki and approval of the Medical Ethics Committee and the competent authorities was obtained. The study was registered at the European Clinical Trial Database (EudraCT 2019-00276-41) and the Dutch Trial Registry (NL8067).
2.1 PatientsPatients were included if they were aged 18 years or older, had a confirmed diagnosis of mCRC or metastatic gastric cancer with a registered indication for trifluridine/tipiracil, an Eastern Cooperative Oncology Group performance status ≤ 1, a body mass index between 18 and 30 kg/m2, and adequate kidney (estimated glomerular filtration rate ≥ 30 mL/min) and liver functions (serum aspartate aminotransferase and alanine aminotransferase ≤ 2.5 upper limit of normal). Patients were excluded if they could not abstain from medications or supplements that could interfere with either trifluridine/tipiracil, metformin or cimetidine (e.g. strong OCT2 or MATE1 inhibitors), had (any type of) diabetes, a known impaired drug absorption or any serious disease interfering with study treatment (e.g. HIV, hepatitis or organ transplant). All patients provided written informed consent.
2.2 Study DesignEligible patients were already on treatment with or started treatment with trifluridine/tipiracil in a dosage of 35 mg/m2 twice a day (b.i.d.) during days 1–5 and 8–12 in 28-day treatment cycles. Upon discretion of the treating physician, reduced doses up to 20 mg/m2 b.i.d. were permitted in the case of toxicity. Phase A in this study (treatment days 1–5) consisted of trifluridine/tipiracil alone (Fig. 2). In Phase B, metformin 500 mg b.i.d. was added to trifluridine/tipiracil (days 8–12), followed by 2 additional days of metformin monotherapy (days 13 and 14). In Phase C, cimetidine 400 mg b.i.d. was combined with trifluridine/tipiracil during days 1–5 of a second treatment cycle. All patients were treated in the sequence of Phase A-B-C. Patients were provided with diaries to accurately record the timing and method of administering trifluridine/tipiracil, metformin and cimetidine during each study period. They were also required to return the used packaging of these medications to ensure proper tracking of drug usage.
Fig. 2
Study overview. PK pharmacokinetic
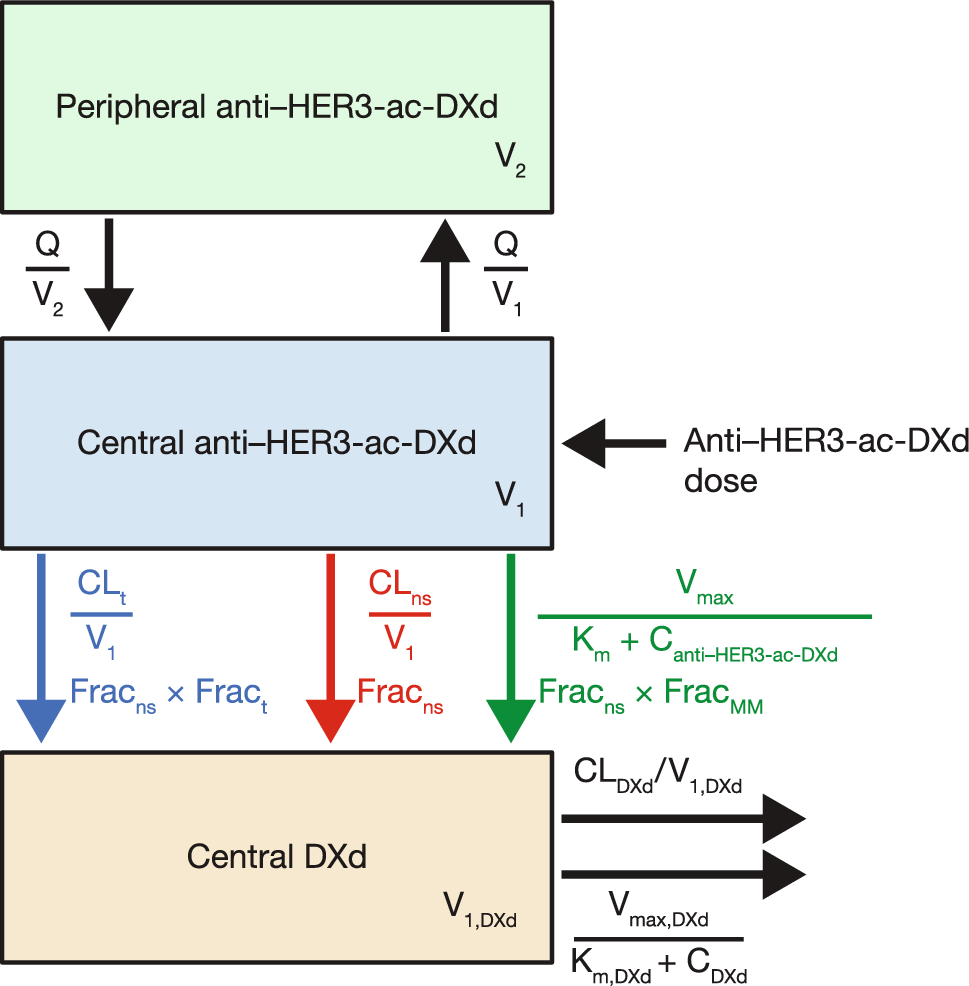
2.3 PharmacokineticsPatients were admitted to the hospital on days 5 and 12 of the first trifluridine/tipiracil treatment cycle and on day 5 of the second treatment cycle for pharmacokinetic blood sampling. On the days of hospital admission, food intake was not permitted between 4 hours prior to and 1 hour after trifluridine/tipiracil administration. Free consumption of beverages was restricted between 1 hour before and 1 hour after trifluridine/tipiracil intake. Blood samples were collected before trifluridine/tipiracil intake and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8 and 12-hour timepoints after trifluridine/tipiracil intake. Two days after the second sampling day (day 14 of the first trifluridine/tipiracil cycle, to ensure a sufficient washout period of trifluridine/tipiracil), an additional blood sample was taken to determine a metformin Ctrough. Blood samples were collected in 4-mL lithium heparin blood collection tubes, processed into plasma within 30 minutes by centrifugation for 10 minutes at 2500–3000g (at 4 °C), and stored at −70 °C until the time of analysis. Trifluridine and metformin plasma concentrations were measured using a validated ultra-performance liquid chromatography-tandem mass spectrometry method. Briefly, trifluridine was extracted from 30-μL aliquots of human lithium heparinised plasma after the addition of 10 μL of internal standard working solution (10 ng/mL of triflurothymidine-13C,15N2 in acetonitrile) and 1.5 mL of ethyl acetate. After centrifugation, the organic phase was evaporated at 60 degrees under a gently stream of nitrogen. The residue was dissolved in an aliquot of 50 µL of water/formic acid (100:0.02, v/v) from which 2 µL was injected onto the high-performance liquid chromatography column. The concentrations were linear over the range of 10.0–1000 ng/mL with a lower limit of quantitation of 10.0 ng/mL. For the metformin analysis, 50 µL of human lithium heparinised plasma was mixed with 400 µL of a 2-mg/L metformin-d6 in methanol solution for protein precipitation. After vortexing and centrifugation, 50 µL of the supernatant was diluted with 800 µL of deionised water, 2 mM ammonium acetate and 0.1% formic acid from which 1 µL was injected into the ultra-performance liquid chromatography system. The concentrations were linear over a range of 0.2–80 mg/L. Pharmacokinetic parameters included exposure defined as the area under the curve from timepoint zero to infinity (AUC0–INF), maximum plasma concentration (Cmax) and the time until maximum plasma concentration (Tmax). The AUC0–INF and Cmax were dose corrected to 35 mg/m2 b.i.d. [pharmacokinetic parameter * (standard dose (35 mg/m2)/administered dose], as trifluridine was found to be dose proportional at the dose range of 40–70 mg/m2/day [15]. All pharmacokinetic parameters were calculated using Phoenix WinNonlin version 8.3 (Certara, Princeton, NJ, USA).
2.4 ToxicityToxicity was assessed during regular outpatient clinics, at baseline and during the pharmacokinetic sampling days, using the Common Terminology Criteria for Adverse Events (version 5.0, National Cancer Institute, Bethesda, MD, USA). All patients were given a diary during the study period and were requested to report any new or ongoing adverse events.
2.5 Statistical AnalysisThe primary endpoint was the relative difference (RD) in exposure to AUC0–INF trifluridine, when used alone and in combination with metformin and cimetidine, respectively. A Bonferroni correction was applied by using an alpha of 0.025 and the calculation of 97.5% confidence intervals. A RD in the area under the curve of at least 30% was considered clinically relevant and the within-patient standard deviation was assumed to be 30% [16]. Based on the above parameters and after rounding to an even number, a total of 18 evaluable patients were required to detect a difference in RD with 80% power. Patients were evaluable if they completed all three phases (per-protocol analysis).
Analyses of the AUC0–INF, the Cmax and the minimum plasma concentration were performed on log-transformed observations, as these were assumed to follow a log-normal distribution [17]. Estimates for the mean differences in (log) area under the curve and Cmax were obtained for the two comparisons separately, and both were analysed using a paired t-test. The mean differences and CIs were then exponentiated to calculate the ratio of the geometric means, which can be interpreted as the RD in percentages. The Tmax was analysed by means of the Wilcoxon signed rank test and described with medians and interquartile ranges. The Ctrough of metformin combined with trifluridine/tipiracil was compared with the Ctrough of metformin monotherapy using a paired t-test on log-transformed data. Toxicity was described as the incidence of toxicity per study phase and was corrected for baseline toxicity. Chi-square tests were used to test for a statistical difference in toxicity between the different study phases.
Comments (0)