
Remember me


Parkinson's disease (PD) is the most common neurodegenerative movement disorder. The characteristic motor symptoms of PD include tremors at rest, rigidity, and bradykinesia. In addition, PD is characterized by a wide variety of non-motor features, including dementia, depression, sleep disturbance, orthostatic hypotension, and psychotic symptoms.[1,2] Moreover, it is pathologically characterized by the progressive death of dopaminergic neurons (DAs) in the substantia nigra pars compacta and the accumulation of cytosolic toxic α-synuclein (SNCA) inclusions known as Lewy bodies.[2,3] Severe dopamine depletion in the midbrain is responsible for the motor symptoms associated with PD, especially bradykinesia, tremors, rigidity, and the loss of postural control. To date, more than 20 genes whose mutations cause autosomal dominant (e.g., SNCA, UCHL1, LRRK2, VPS35), autosomal recessive (e.g., PINK1, PARK7, ATP13A2, FBXO7), and X-linked (RAB39B) monogenic PD have been found,[4] with related phenotypes ranging from idiopathic PD-like to young-onset Parkinsonism, with or without atypical motor and non-motor features. Familial monogenic PD, with mutations in identified genes such as SNCA, LRRK2, and PINK1, accounts for only 3–5% of PD cases.[5–7] However, the etiology of sporadic PD remains largely unclear. The current view is that PD is a complex disorder occurring due to the interplay among genetic, environmental, nutritional, and other factors, together with aging.[8] Although PD is still an incurable, progressive disease, treatment involves pharmacologic and non-pharmacologic approaches, substantially improving quality of life and functional capacity.[9]
Transcription is the first step in gene expression. It is modulated through interactions between a series of transcription factors and typical transcription factor-binding sites, mainly located in gene promoter regions. Developmental studies have suggested that transcription factors play critical roles in the specification, development, and survival of DAs in the substantia nigra throughout life.[10,11] Typical DA transcription factors determine the DA expression phenotype and survival by triggering specific intracellular transcription cascades,[10,12] including engrailed homeobox 1 and 2 (EN1 and EN2), forkhead box A1 and 2 (FOXA1 and FOXA2), LIM homeobox transcription factor 1 alpha and beta (LMX1A and LMX1B), nuclear receptor subfamily 4 group A member 2 (NR4A2), orthodenticle homeobox 2 (OTX2), paired like homeodomain 3 (PITX3), gastrulation brain homeobox 2 (GBX2), GLI family zinc finger 1 (GLI1), Msh homeobox 1 (MSX1), neurogenin 2 (NGN2), early B cell factor transcription factor 1 (EBF1), SRY-box transcription factor 6 (SOX6), and SRY-box transcription factor 2 (SOX2). Genetic variants in some genes encoding certain dopaminergic transcription factors have been suggested to be risk factors for PD.[13–16] However, no comprehensive genetic analyses of these typical dopaminergic transcription factors in patients with PD have been performed.
In this study, we combined genes involved in the regulation of midbrain DA differentiation based on the associated Gene Ontology (GO) category (GO: 0071542) with genes summarized in recent reviews.[10,12] Then, we included 16 targeted transcription factor genes. Variants of EN1, EN2, FOXA1, FOXA2, LMX1A, LMX1B, NR4A2, OTX2, PITX3, GBX2, GLI1, MSX1, NGN2, EBF1, SOX6, and SOX2 and their potential association with PD were determined using whole-exome sequencing (WES) and whole-genome sequencing (WGS) based on cohorts of Chinese patients with PD.
Methods SubjectsPatients of Chinese origin with PD from Xiangya Hospital, Central South University, and other cooperating centers of the Parkinson's Disease and Movement Disorders Multicenter Database and Collaborative Network in China (http://pd-mdcnc.com/) were enrolled between October 2006 and December 2020. All patients were diagnosed by two expert neurologists according to the clinical diagnostic criteria of the United Kingdom Parkinson's Disease Society Brain Bank[17] or the International Parkinson and Movement Disorders Society.[18] Unrelated individuals without the disorder were recruited from local communities as controls. Mutations in known pathogenic genes associated with PD were excluded in all subjects, according to the principles mentioned in our previous study.[7] The WES group included patients with a family history of PD or an onset age less than or equal to 50 years with sporadic PD and the controls. The WGS group comprised patients with an onset age greater than 50 years with sporadic PD and the controls. The participant demographic characteristics are listed in Supplementary Table 1, https://links.lww.com/CM9/B599.
This study was approved by the relevant oversight committees and institutional review boards and the Ethics of Committee of Xiangya Hospital, Central South University in this study approved the protocol (No. 202004049). Written consent was obtained from all individuals.
WES, WGS, and bioinformatic analysesGenomic deoxyribonucleic acid (DNA) was extracted from peripheral blood leukocytes. WES, WGS, and quality control were performed, as described previously.[7,19] Specifically, we conducted WES using SureSelect Human All Exon Kit V6 (Agilent Technologies, Santa Clara, CA, USA) to capture the whole-exome DNA and prepare sample libraries, followed by paired-end 2 × 150 bp sequencing with an Illumina HiSeq X10 Sequencing System (Illumina, San Diego, CA, USA). Moreover, we conducted WGS using an Illumina Nova sequencing platform in paired-end 2 × 150 bp mode. Paired-end read alignment (GRCh37/hg19), duplicate read removal, variant calling (single-nucleotide variants and small insertion or deletions), and variant annotations were performed using the Burrows–Wheeler Aligner,[20] Picard tools (http://broadinstitute.github.io/picard/), Genome Analysis Toolkit,[21] and ANNOVAR[22] programs, respectively.
For individual and variant quality control, PLINK v1.90 (https://zzz.bwh.harvard.edu/plink/) was used.[23] Individuals were excluded if they showed ambiguous sex, low genotype call rates (missing rate >5%), deviating heterozygosity/genotype calls (±3 standard deviations), or cryptic relatedness (identity by descent >0.15). Variant quality control was accomplished through the removal of low-quality genotypes (Phred-scaled genotype quality score below 30, allele depth [AD] below 10, and read depth [DP] below 30 for WES data; Phred-scaled genotype quality score below 15, AD below 2, and DP below 5 for WGS data) and variants with low call rates (missing rate >5%) or departure from the Hardy–Weinberg equilibrium (P <0.001). Next, principal component analysis for population stratification was performed with samples from the 1000 Genomes Project using linkage equilibrium pruned variants (r2 <0.1), and the visually inspected outliers suggesting a non-Chinese origin were excluded.[24] Next, the principal component variables for each sample were obtained using principal component analysis (PCA) in PLINK. The top five principal components of ancestry were included as covariates in the following analysis. All variants in the coding regions and 2 kb up- or down-stream of the coding regions of the 16 genes involved in the developmental differentiation of midbrain DAs were included in this study [Table 1]. The corresponding quality parameters of the targeted regions were presented in Supplementary Table 2, https://links.lww.com/CM9/B599.
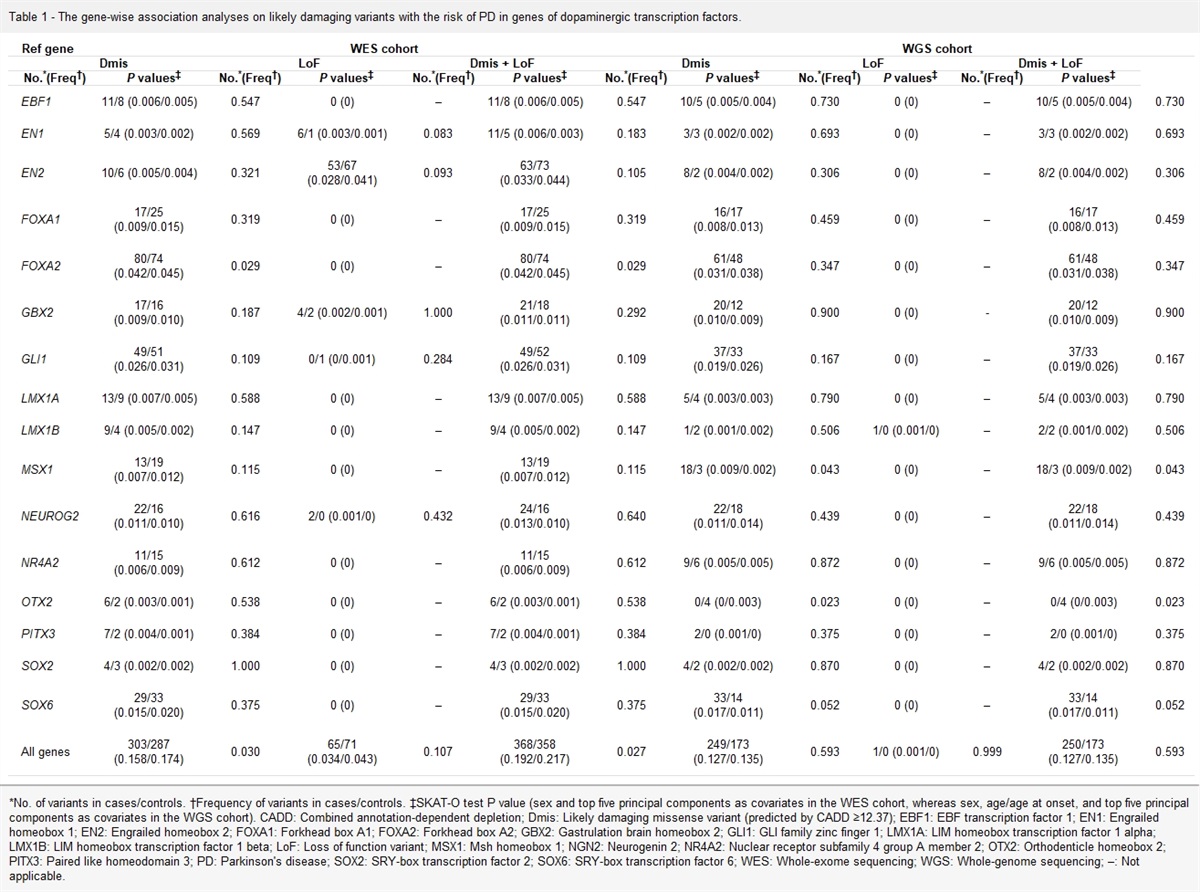
Table 1 - The gene-wise association analyses on likely damaging variants with the risk of PD in genes of dopaminergic transcription factors. Ref gene WES cohort WGS cohort Dmis LoF Dmis + LoF Dmis LoF Dmis + LoF No. *(Freq †) P values ‡ No. *(Freq †) P values ‡ No. *(Freq †) P values ‡ No. *(Freq †) P values ‡ No. *(Freq †) P values ‡ No. *(Freq †) P values ‡ EBF1 11/8 (0.006/0.005) 0.547 0 (0) – 11/8 (0.006/0.005) 0.547 10/5 (0.005/0.004) 0.730 0 (0) – 10/5 (0.005/0.004) 0.730 EN1 5/4 (0.003/0.002) 0.569 6/1 (0.003/0.001) 0.083 11/5 (0.006/0.003) 0.183 3/3 (0.002/0.002) 0.693 0 (0) – 3/3 (0.002/0.002) 0.693 EN2 10/6 (0.005/0.004) 0.321 53/67 (0.028/0.041) 0.093 63/73 (0.033/0.044) 0.105 8/2 (0.004/0.002) 0.306 0 (0) – 8/2 (0.004/0.002) 0.306 FOXA1 17/25 (0.009/0.015) 0.319 0 (0) – 17/25 (0.009/0.015) 0.319 16/17 (0.008/0.013) 0.459 0 (0) – 16/17 (0.008/0.013) 0.459 FOXA2 80/74 (0.042/0.045) 0.029 0 (0) – 80/74 (0.042/0.045) 0.029 61/48 (0.031/0.038) 0.347 0 (0) – 61/48 (0.031/0.038) 0.347 GBX2 17/16 (0.009/0.010) 0.187 4/2 (0.002/0.001) 1.000 21/18 (0.011/0.011) 0.292 20/12 (0.010/0.009) 0.900 0 (0) - 20/12 (0.010/0.009) 0.900 GLI1 49/51 (0.026/0.031) 0.109 0/1 (0/0.001) 0.284 49/52 (0.026/0.031) 0.109 37/33 (0.019/0.026) 0.167 0 (0) – 37/33 (0.019/0.026) 0.167 LMX1A 13/9 (0.007/0.005) 0.588 0 (0) – 13/9 (0.007/0.005) 0.588 5/4 (0.003/0.003) 0.790 0 (0) – 5/4 (0.003/0.003) 0.790 LMX1B 9/4 (0.005/0.002) 0.147 0 (0) – 9/4 (0.005/0.002) 0.147 1/2 (0.001/0.002) 0.506 1/0 (0.001/0) – 2/2 (0.001/0.002) 0.506 MSX1 13/19 (0.007/0.012) 0.115 0 (0) – 13/19 (0.007/0.012) 0.115 18/3 (0.009/0.002) 0.043 0 (0) – 18/3 (0.009/0.002) 0.043 NEUROG2 22/16 (0.011/0.010) 0.616 2/0 (0.001/0) 0.432 24/16 (0.013/0.010) 0.640 22/18 (0.011/0.014) 0.439 0 (0) – 22/18 (0.011/0.014) 0.439 NR4A2 11/15 (0.006/0.009) 0.612 0 (0) – 11/15 (0.006/0.009) 0.612 9/6 (0.005/0.005) 0.872 0 (0) – 9/6 (0.005/0.005) 0.872 OTX2 6/2 (0.003/0.001) 0.538 0 (0) – 6/2 (0.003/0.001) 0.538 0/4 (0/0.003) 0.023 0 (0) – 0/4 (0/0.003) 0.023 PITX3 7/2 (0.004/0.001) 0.384 0 (0) – 7/2 (0.004/0.001) 0.384 2/0 (0.001/0) 0.375 0 (0) – 2/0 (0.001/0) 0.375 SOX2 4/3 (0.002/0.002) 1.000 0 (0) – 4/3 (0.002/0.002) 1.000 4/2 (0.002/0.002) 0.870 0 (0) – 4/2 (0.002/0.002) 0.870 SOX6 29/33 (0.015/0.020) 0.375 0 (0) – 29/33 (0.015/0.020) 0.375 33/14 (0.017/0.011) 0.052 0 (0) – 33/14 (0.017/0.011) 0.052 All genes 303/287 (0.158/0.174) 0.030 65/71 (0.034/0.043) 0.107 368/358 (0.192/0.217) 0.027 249/173 (0.127/0.135) 0.593 1/0 (0.001/0) 0.999 250/173 (0.127/0.135) 0.593*No. of variants in cases/controls. †Frequency of variants in cases/controls. ‡SKAT-O test P value (sex and top five principal components as covariates in the WES cohort, whereas sex, age/age at onset, and top five principal components as covariates in the WGS cohort). CADD: Combined annotation-dependent depletion; Dmis: Likely damaging missense variant (predicted by CADD ≥12.37); EBF1: EBF transcription factor 1; EN1: Engrailed homeobox 1; EN2: Engrailed homeobox 2; FOXA1: Forkhead box A1; FOXA2: Forkhead box A2; GBX2: Gastrulation brain homeobox 2; GLI1: GLI family zinc finger 1; LMX1A: LIM homeobox transcription factor 1 alpha; LMX1B: LIM homeobox transcription factor 1 beta; LoF: Loss of function variant; MSX1: Msh homeobox 1; NGN2: Neurogenin 2; NR4A2: Nuclear receptor subfamily 4 group A member 2; OTX2: Orthodenticle homeobox 2; PITX3: Paired like homeodomain 3; PD: Parkinson's disease; SOX2: SRY-box transcription factor 2; SOX6: SRY-box transcription factor 6; WES: Whole-exome sequencing; WGS: Whole-genome sequencing; –: Not applicable.
We selected rare variants obtained from datasets with a minor allele frequency less than or equal to 0.01. In contrast, common variants were defined as those with a minor allele frequency greater than 0.01. The minor allele frequency was based on the East Asian population of the exome sequencing data (8624 East Asian individuals) and genome sequencing data (780 East Asian individuals) from the GnomAD (v2.0.2, downloaded on 2018-08-03) and ExAc databases (v1.0, downloaded on 2018-08-14; 4312 East Asian individuals). Rare variants were divided into five subgroups: synonymous, missense, likely damaging missense, loss-of-function, and likely damaging missense plus loss-of-function variants groups. Of these, likely damaging missense variants were identified based on a combined annotation-dependent depletion (CADD) C-score of no less than 12.37, similar to previous studies.[25] In previous studies, a CADD score >12.37 represented a subset with the top 2% most damaging of all possible missense changes in the genome, which was enriched for known pathogenic variants.[25,26]
We conducted Sanger sequencing on several random rare damaging variants. The results showed that these variants are correct [Supplementary Figure 1, https://links.lww.com/CM9/B599], and the primer sequences for Sanger sequencing were listed in Supplementary Table 3, https://links.lww.com/CM9/B599.
Statistical analysesGene-level burden analyses were conducted for the two PD cohorts. All rare, likely damaging missense and loss-of-function variants were identified as putative damaging variants to test the accumulated association of these variants in each gene with PD. The optimized sequence kernel association test (SKAT-O)[27] was used for the gene-based analysis of rare variants using the SKAT R package (https://cran.r-project.org/web/packages/SKAT/index.html); it was performed on five variant subgroups of individual genes and the entire gene set. First, we conducted the gene-wise association analyses on likely damaging variants with the risk of PD in genes of dopaminergic transcription factors and the whole gene set. Next, we conducted the gene-wise association analyses on likely damaging variants with disease severity in genes of dopaminergic transcription factors and the whole gene set. In this case, disease severity was classified according to Hoehn and Yahr stages, among which stages 1–2.5 were classified as early stage PD, and stages 3 and above were classified as late-stage PD.
PLINK was used for logistic regression analysis of single common variants.[23] All common variants were included in the analyses, regardless of where they were located or whether they affected the amino acid change. Sex and the first five principal components of ancestry were included as covariates in the WES cohort. In contrast, age/age at onset (age of control and age at onset of PD patients), sex, and the first five principal components of ancestry were included as covariates in the WGS cohort. The significance threshold was adjusted using the Bonferroni correction in the rare variant analysis, as 16 independent tests were conducted. For the common variants, we included 72 and 1730 common variants in the WES and WGS cohorts, respectively. However, considering that the variants were inter-dependent, we used r2 <0.1 as the cut-off to perform the independent tests in our analysis, which included 39 tests for the WES cohort and 160 tests for the WGS cohort. Therefore, Bonferroni's correction was conducted in the single-variant analysis of common variants in the WES and WGS cohorts.
To evaluate the statistical power of our study, we conducted 1000 SKAT simulations on targeted regions in both cohorts. We assumed the prevalence of PD to be 0.107% and 0.428% for the WES and WGS cohorts, respectively, considering their different onset ages.[28] The length of targeted regions was set to the sum of 16 dopaminergic transcription factor genes (22.2 kb) or the average length of those 16 genes (1.4 kb) for the gene-set simulations or single gene simulations. Moreover, the minor allele frequency cut-off for causal variants was set to 0.00035.[25]
Results Rare variants identified in WES and WGS cohortsThe WES cohort included 1917 unrelated patients with familial or sporadic early onset PD and 1652 controls. We detected 308 rare protein-altering variants (183 likely damaging missense among 295 missense variants and 13 loss-of-function variants) in 16 individual genes [Supplementary Table 4, https://links.lww.com/CM9/B599]. Gene-based burden analyses of these rare variants showed that FOXA2 was significantly associated with PD (likely damaging missense variants: Frequency of variants (Freq)cases/Freqcontrols = 0.042/0.045, P = 0.029), whereas other genes did not show any association with PD in any variant subgroup [Table 1]. Furthermore, the dopaminergic transcription factor gene set was associated with PD (likely damaging missense variants: Freqcases/Freqcontrols = 0.158/0.174, P = 0.030; likely damaging missense and loss-of-function variants: Freqcases/Freqcontrols = 0.192/0.217, P = 0.027). However, after Bonferroni correction, none of the 16 gene variants were significantly enriched in controls, compared with their prevalence in patients with familial or sporadic early-onset PD [Table 1, Supplementary Table 5, https://links.lww.com/CM9/B599]. Moreover, none of the 16 gene variants were significantly enriched in early- or late-stage PD [Supplementary Table 6, https://links.lww.com/CM9/B599].
The WGS cohort included 1962 unrelated patients with sporadic late-onset PD and 1279 controls. In total, 208 rare protein-altering variants were identified [Supplementary Table 4, https://links.lww.com/CM9/B599]. The likely damaging missense variants in OTX2 were found to be suggestively enriched in controls (Freqcases/Freqcontrols = 0/0.003, P = 0.023). In contrast, the likely damaging missense variants of MSX1 were found to be suggestively enriched in PD (Freqcases/Freqcontrols = 0.009/0.002, P = 0.043). The remaining genes were not associated with PD in any variant subgroup [Table 1], including variants in all genes encoding dopaminergic transcription factors. Nevertheless, following Bonferroni correction, none of these genes were significantly associated with sporadic late-onset PD [Table 1]. Although single gene analysis was relatively weak in evaluating the role of a single gene, the statistical power of our analysis, which used the entire gene set as the object, was somewhat improved, and still, no statistically significant difference was indicated. Similarly, none of the 16 gene variants were significantly enriched in early or late-stage PD [Supplementary Table 6, https://links.lww.com/CM9/B599].
Common variants identified in WES and WGS cohortsCommon variants in the 16 genes were also examined in both cohorts. Overall, 72 common variants were found in the WES cohort [Supplementary Table 7, https://links.lww.com/CM9/B599], and 1730 were identified in the WGS cohort [Supplementary Table 8, https://links.lww.com/CM9/B599]. We found four variants associated with sporadic or familial early-onset PD, including one in each of LMX1A, MSX1, PITX3, and SOX6. Furthermore, we identified 92 variants suggestively associated with sporadic late-onset PD. However, after using single-variant logistic association analyses and Bonferroni correction, we did not identify any significant associations between these common variants and PD.
DiscussionThe DA-specific transcription factors include EN1, EN2, FOXA1, FOXA2, LMX1A, LXM1B, NR4A2, OTX2, PITX3, GBX2, GLI1, MSX1, NGN2, EBF1, SOX6, and SOX2. GBX2, OTX2, FOXA1, FOXA2, GLI1, EN1, and EN2, which are mainly involved in DA neuron induction; NGN2, MSX1, LMX1A, and LMX1B, mainly involved in DA neuron specification; and NR4A2 and PITX3, mainly involved in DA neuron differentiation and maturation, whereas EBF1, SOX6, and SOX2 are involved in DA migration.[12] Several functional studies have demonstrated that these transcription factors are important for post-mitotic DA development and maintenance.[11] For example, NR4A2 was proven to be strictly coupled with neurotransmitter synthesis. A reduction in NR4A2 expression, induced by elevated SNCA cellular levels, has an invaluable role in the induction of DA dysfunction in the early stages of PD.[29] EN1 and EN2 act on at least two steps of the differentiation process of midbrain DAs and are essential for DA survival.[10] Furthermore, PITX3 is highly expressed in midbrain DAs, which are susceptible to environmental or endogenous stress.[30] Additionally, FOXA2 encodes FOXA2 protein, which regulates DA generation and differentiation. Previous studies have revealed that aging Foxa2+/- mice spontaneously develop Parkinsonism; thus, FOXA2 is considered essential to regenerative medicine.[30] Moreover, OTX2 acts as a gap gene in the early development of the anterior part of the central nervous system, encoding the OTX2 protein expressed in the DAs of the ventral tegmental area.[30]
Although the functions of these transcription factors have been extensively studied,[31–35] genetic research on typical dopaminergic transcription factors is insufficient and controversial.[13–16,36,37] In particular, genetic studies investigating the role of FOXA2, OTX2, GBX2, GLI1, NGN2, EBF1, SOX6, and SOX2 variants in patients with PD have not been conducted. It is generally thought that transcription is the first step in gene expression. It is modulated through interactions between a series of transcription factors and typical transcription factor-binding sites in gene promoter regions. Typical DA transcription factors play important roles in DA expression and survival by controlling specific pathways. Therefore, we assumed that missense, loss-of-function, or gain-of-function variants in these DA transcription factors could compromise their transcriptional activation and potentially disrupt DA development and maintenance, which might trigger pathological changes that lead to PD. In this study, we identified variants in 16 typical dopaminergic transcription factors among patients with PD and control subjects from China. We comprehensively analyzed the association between the variants and PD risk. Our results indicated that the variants in these transcription factors were not significantly associated with the risk of either familial/sporadic early-onset or sporadic late-onset PD in this Chinese cohort. In humans, MSX1 variants have been related to tooth agenesis, orofacial deformities, and nail dysplasia.[38] A previous study revealed that no genetic variations in the coding region of the MSX1 gene were identified in another Chinese cohort.[39] In our study, it should be noted that the rare, likely damaging missense variants identified in MSX1 were suggestively associated with sporadic late-onset PD. Although significance did not pass the Bonferroni correction, this result provides sufficient interest to encourage further studies of this gene in PD. In a German cohort, previous studies did not identify association signals in the coding region of the OTX2 gene.[40] Moreover, the rare, likely damaging missense variants found in FOXA2 or OTX2 were suggestively enriched in controls, compared with their prevalence in sporadic early-onset/familial PD or sporadic late-onset PD, respectively, which should be interpreted cautiously.
Our study did not find any association between these 16 typical dopaminergic transcription factors and PD, which was partly in line with the results reported in previous GWAS summary data[41] [Supplementary Tables 7 and 8, https://links.lww.com/CM9/B599]. Besides, we did not identify any association between these 16 typical dopaminergic transcription factors and the severity of PD motor syndrome. Furthermore, one review showed that rs2281983 and rs4919621 SNPs in PITX3 are related to early-onset PD risk in Caucasians but not in other cohorts.[42] Similarly, we did not report an association between these SNPs and PD risk in the Chinese population. A previous study found a strong and reproducible association of the PITX3 promoter SNP rs3758549 with PD,[40] but we did not find this association in our population (P = 0.656). It indicates that ethnic backgrounds should be considered in association studies and should thus be of sufficient interest to encourage further studies of these genes in other ethnic groups.
We acknowledge that our study had some limitations. First, although we had a relatively large case-control cohort, the statistical power of the analysis was still limited for gene-based rare variant analysis (approximately 22% and 21% in the WES and WGS cohorts, respectively). However, a gene-set-based analysis could increase the power (82% or 78%). We are anticipating studies using larger and well-stratified cohorts to improve statistical power. Second, the expression of transcription factors and the transcriptomes of downstream target genes regulated by transcription factors were not investigated in our cohort. Future studies on transcriptome changes in target gene
Comments (0)