
Remember me


Pancreatic cancer (PC) is a highly malignant disease with a poor prognosis. The latest National Comprehensive Cancer Network guidelines (2019) indicate that its 5-year survival rate is approximately 9%, and the second most deadly tumor is expected to be PC by 2030.[1] PC has such feature not only because of its occult nature but also its traits of early metastasis and high invasiveness; thus, many advanced patients miss the window for surgical intervention. Therefore, chemotherapeutic treatment has become vital for prolonging patient survival. However, despite the severe adverse drug response, the first-line drugs remain primarily 5-fluorouracil (5-FU)-based Folinic acidorouracil, Irinotecan, Oxaliplatin (FOLFIRINOX) and gemcitabine-based regimens.[1]Although many studies have attempted to clarify the cause of chemotherapy's poor effect on PC by evaluating its subtypes and molecular mechanisms,[2] the chemoresistance mystery remains. At the 2018 American Society of Clinical Oncology meeting, a randomized controlled trial for PRODIGE 24/CCTG PA.6 was presented, confirming that compared with standard gemcitabine adjuvant therapy, moderate FOLFIRINOX adjuvant therapy increased the average survival time of patients with PC to 20 months, prolonged disease-free and metastasis-free survival, and showed safety and tolerability.[3] In addition, albumin-bound paclitaxel in combination with gemcitabine, erlotinib in combination with gemcitabine, and cisplatin in combination with gemcitabine are being used with improved effects.[4]
Continued research in PC has shown metabolism to be a primary and direct way that PC cells achieve chemoresistance. Just as hypovascularity and high fibrosis reduce drug concentration in cancer tissues,[5] the metabolic characteristics of PC that antagonize chemotherapeutic drugs also weaken chemotherapy's effects.[5]
This review summarizes the current understanding of metabolism's contribution to chemotherapy resistance and the metabolic molecules and key pathways that offer therapeutic targets for reversing chemoresistance in PC.
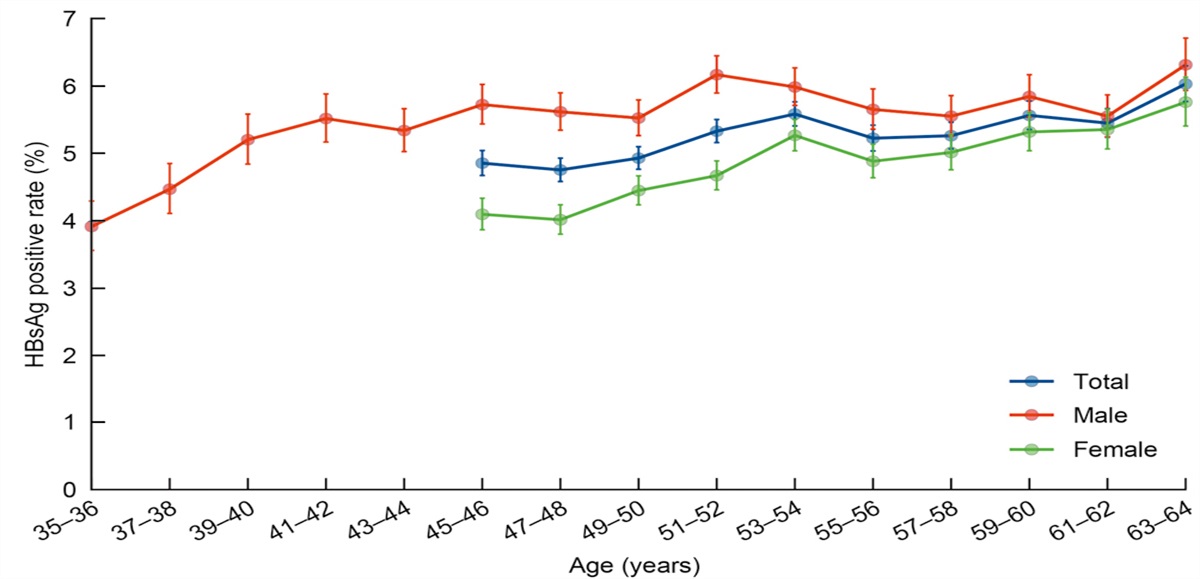
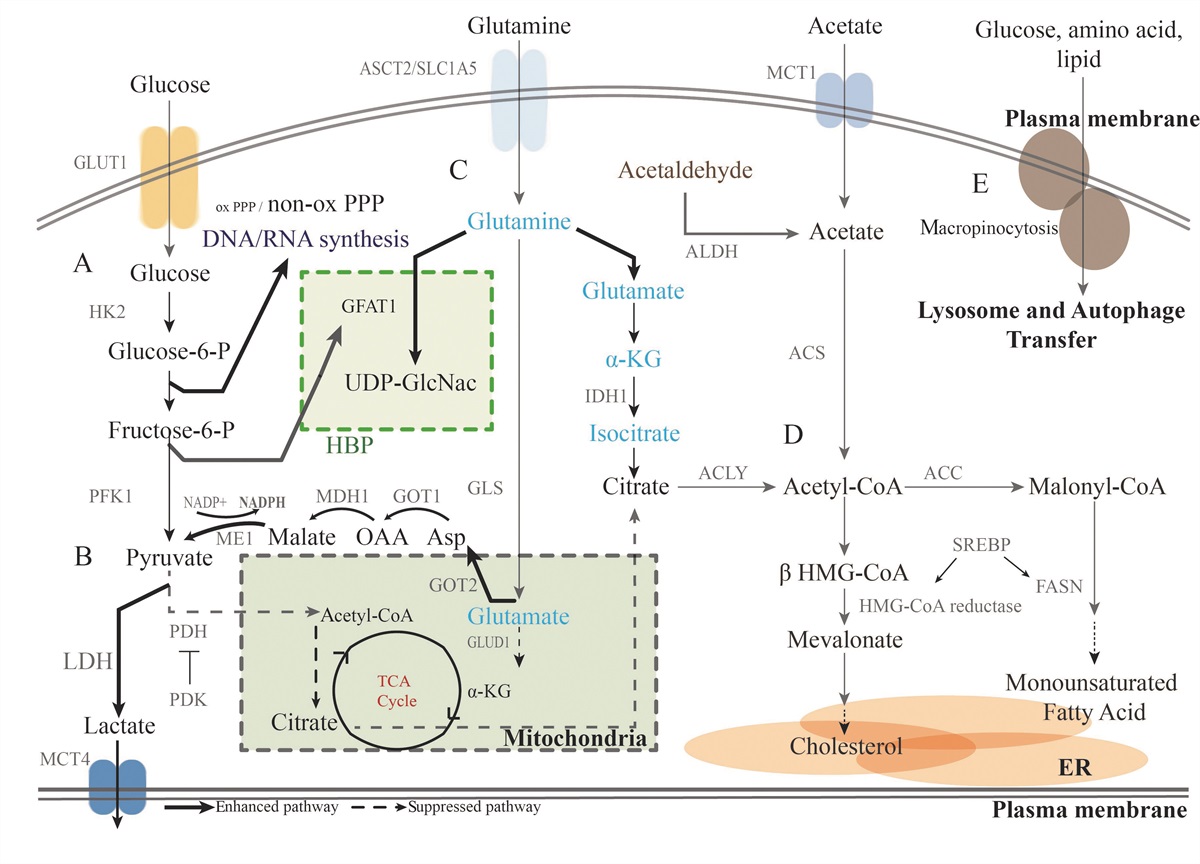
Pathway and Enzyme Reprograming in the PC Metabolic ProcessResearch on the mechanism of pancreatic ductal adenocarcinoma (PDAC) initiation and development suggests induction of metabolic reprograming, with different patterns of enzyme expression leading to specific modes of metabolism for glucose, amino acids, and lipids through rewired pathways [Figure 1].[6] Understanding how this process contributes to chemoresistance has revealed potential therapeutic targets for PC.
 Figure 1:
Figure 1: Metabolic reprograming in PC. (A) Glucose metabolism is reprogramed in PDAC. By upregulating GLUT1 expression, PC drives glycolysis through multiple glycolytic enzymes. In addition, glycolytic intermediates are forced into biosynthetic pathways, such as the non-oxidative PPP and the HBP, which play important roles in generating sufficient products for proliferation and survival. (B) Pyruvate tends to be shunted for the generation of lactate by increased LDH expression, while the TCA cycle is shut down by the high expression of PDK, which inhibits PDH. (C) Glutamine plays several key roles in PC cells, including participating in the HBP to synthesize sufficient UDP-GlcNac and being processed into citric acid for fatty acid synthesis. Glutamine tends to be transformed to pyruvate by a series of enzyme activations rather than moved into the TCA cycle because of the inhibition of GLUD1. (D) Acetyl-CoA has several sources in PDAC cells. (E) Direct macropinocytosis and autophagy also provide glucose, amino acids, and FAs to cancer cells, and lysosomes play an important role in this process. ACLY: ATP-citrate lyase; ACS: Acetyl-CoA synthetase; Acetyl-CoA: Acetyl-Coenzyme A; ALDH: Aldehyde dehydrogenase; ATP: Adenosine triphosphate; ER:Endoplasmic reticulum; FASN: FA synthase; GFAT1:Glucosamine-fructose-6-phosphate aminotransferase 1; GLS: Glutaminase; GLUT1: Glucose transporter type 1; GOT1: Aspartate aminotransferase 1; HBP: Hexosamine biosynthesis pathway; HK2: Hexokinase-2; HMG-CoA: 3-Hydroxy-3-methylglutaryl-coenzyme A; LDH: Lactate dehydrogenase; LDH-A: Lactate dehydrogenase A; ME1: Malic enzyme 1; MDH1: Malate dehydrogenase; OAA: Oxaloacetate; PC: Pancreatic cancer; PDAC: Pancreatic ductal adenocarcinoma; PDH: Pyruvate dehydrogenase; PDK: Pyruvate dehydrogenase kinase; PFK1: phospofructokinase 1; PPP: Pentose phosphate pathway; SREBP: Sterol regulatory element-binding protein; TCA: Tri-cyclic acid; UDP-G1cNac: Uridine diphosphate N-acetylglucosamine. NADPH; GLUD; ASP; a-KG; MCT4; SLC1A5; IDH1; ACC; SREBP
Key enzymes and pathway reprograming in glycolysisPrevious studies have shown that PC cells have high rates of glucose uptake and lactic acid fermentation, thereby promoting rapid biosynthesis and adenosine triphosphate (ATP) synthesis; this is termed the Warburg effect.
Glucose uptake is also increased in PDAC cells. Glucose transporter type 1 (GLUT1), whose expression is upregulated by KRAS mutation, is a key transporter in glucose uptake in PDAC, and its expression is an independent prognostic factor.[7] GLUT1 has been shown to induce chemoresistance in malignant tumors, although the exact mechanism underlying this effect has not yet been clarified.[8] In addition, PDAC cells can obtain sugar through Ras-mediated autophagy and lysosomal transfer.[9] Hydroxychloroquine (HCQ) shows the ability to block lysosome acidification and has been used in combination with gemcitabine for the treatment of PDAC in clinical trials.[10]
The glycolytic process in PDAC cells is significantly reprogramed by oncogenes such as KRAS and c-myc, which shuttle glucose away from the traditional tri-cyclic acid (TCA) cycle into other branch pathways.[11] Hexokinase-2 (HK2), an important enzyme in the first step of glycolysis, is one of the most highly expressed molecules in PDAC metastases suggesting a connection between HK2 and PC aggression.[12]
In PDAC, glucose flows into the biosynthetic pathways. The non-oxidative pentose phosphate pathway (PPP) is activated by the transcriptional upregulation of enzymes which increases de novo nucleotide biosynthesis. This reprograming competitively antagonizes the concentration of nucleoside analogs in cells leading to chemoresistance to gemcitabine or 5-FU.[13] Inhibitors of non-oxidative PPP, such as leflunomide, have shown the potential to antagonize the chemoresistance of PDAC and triple-negative breast cancer (TNBC).[14] The hexosamine biosynthesis pathway (HBP) is also reprogramed via the upregulation of glucosamine-fructose-6-phosphate aminotransferase 1 (GFAT1), the rate-limiting enzyme in the HBP.[15] High expression of GFAT1 in PDAC predicts a poor prognosis, while GFAT1 inhibitors can induce tumor regression, indicating further possibilities for developing cancer chemotherapies.[16]
In PDAC, glucose is converted into pyruvate through glycolysis, which has two major fates in tumor cells. First, pyruvate is converted to lactate via lactate dehydrogenase (LDH), which is the most effective way to maintain the NAD+ level needed for high-rate cell proliferation of PDAC.[17] Second, pyruvate is converted into acetyl-Coenzyme A (acetyl-CoA) in the mitochondria via the pyruvate dehydrogenase (PDH) complex to enter the TCA cycle. Several factors are produced under anaerobic conditions or KRAS activation to inhibit PDH and decrease the glucose flux into the TCA cycle.[18] Therefore, tumor cells greatly promote the conversion of pyruvate into lactate, with LDH playing a key role in this process.
LDH is a homo- or heterotetrameric enzyme consisting of two different subunits encoded by the highly related genes LDH-A and LDH-B. LDH-A favors the conversion of pyruvate into lactate, and is greatly elevated in PC and significantly correlated with poor prognosis and chemotherapy resistance,[19] and KRAS, epidermal growth factor receptor (EGFR), c-Myc, and hypoxia inducible factor-1 (HIF-1) have been found to upregulate LDH-A.[20] An inhibitory acetylation at lysine 5 of LDH-A was reduced in PCs, likely explaining the LDH-A activation in PCs.
Lactate is the main product of glycolysis in PDAC, and when overproduced, it can diffuse into the extracellular environment via the monocarboxylate transporter (MCT-1) to then be taken up by normoxic cancer cells through MCT-1 for use in oxidative metabolism, thereby saving glucose for hypoxic cancer cells. MCT is, therefore, a potential target for chemotherapy based on metabolism. Specifically, AZD3965 shows great potential as an MCT-inhibiting cancer treatment when combined with chemotherapy.[21]
Key enzyme and pathway reprograming in amino acid metabolismAmino acid metabolism is also reprogramed in PDAC, using a network between glycolysis and amino acid synthesis.
Glutamine (Gln) is a non-essential and most abundant amino acid in tumor circulation, as well as a major carbon and nitrogen source. Accordingly, high Gln metabolism activity is a key feature of PDAC both in vivo and in vitro, and it is used to maintain redox balance.[22]
The transport of Gln into PDAC cells is increased. Oncogenes such as C-myc upregulate the Gln transporter alanine serine cysteine, preferring transporter 2 (ASCT2) and key glutaminolytic enzymes.[23] ASCT2 is also a key enzyme in Gln transport in PC, leading to the accumulation of Gln in PDAC tumors.[24] Several studies have indicated ASCT2 as a potential therapeutic target in chemoresistant cancers.[25]
Gln functions in amino acid metabolism by first generating reducing equivalents of nicotinamide adenine dinucleotide phosphate (NADPH) in a pathway driven by oncogenic KRAS.[22] Gln must then be catabolized via glutaminase (GLS) into glutamate (Glu) before it can enter the TCA cycle.[26] In PDAC cells, glutamate is ultimately converted into aspartate and released into the cytoplasm, generating oxaloacetate, which is then used for amino acid synthesis by cytoplasmic malic enzyme 1 (ME1). NADPH is synthesized in this process, increasing the NADPH/NADP+ ratio and allowing PDAC cells to maintain redox balance and proliferate.[27] ME1 is the key enzyme that converts malate into pyruvate, thereby completing the amino acid breakdown process and generating carbon cycle fuel in PDAC cells; thus, malate is a potential target for therapy. Novel research has shown that ME1 increases PPP flux by forming physiological complexes with 6-phosphogluconate dehydrogenase leading to glycolysis crosstalk between tumor cells.[28] Downregulation of ME1 expression is effective in sensitizing chemoresistant cancer cells. Gln-derived glutamate is used for glutathione biosynthesis, which is a principal component of cellular redox balance.[29]
GLS, the key enzyme that converts Gln to Glu, has attracted attention as its silencing severely impairs reactive oxygen species (ROS) scavenging systems and causes severe ROS-mediated cytotoxicity, providing a potential target for cancer treatment.[26] A novel study showed that glutaminolysis is important for the survival of PDAC cells that show tolerance to nutrient starvation or chemotherapy and that the use of a combination of acetyl-CoA carboxylase and GLS inhibitors showed good treatment potential for chemoresistant PDAC.[30]
Key enzyme and pathway reprograming in lipid metabolismActivated lipid synthesis is essential for cancer cells. Lipids such as those in phospholipid bilayers are fundamental structural components in cancer cell proliferation, and cancer cells generally synthesize the majority of their non-essential fatty acids (FAs) de novo; however, oncogenic KRAS activation suggests a shift in the origin of FA pools in cancer cells. As a KRAS-driven tumor, PC shows abnormal lipid metabolism, which plays a key role in disease progression.[31]
Generated from citrate by ATP-citrate lyase, cytoplasmic acetyl-CoA has two major fates. First, acetyl-CoA can be converted into malonyl-CoA and then bound to the acyl-carrier protein domain of FA synthase (FASN) to produce monounsaturated palmitic acids. Second, it can be used to synthesize cholesterol via the mevalonate pathway, with 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase as the rate-limiting enzyme.[32] In addition to the de novo synthesis of cholesterol, cells can increase cholesterol levels through receptor-mediated endocytosis of low-density lipoproteins (LDLs). Both LDL receptors and HMG-CoA reductase are targets of Sterol regulatory element-binding protein-2 (SREBP-2), which is significantly overexpressed in KRAS-activated PDAC, leading to increased cholesterol intake and synthesis.[33]
Cancer cells form a tremendous network of several metabolic pathways [Figure 1]. The first essential molecule in FA generation is pyruvate, which is involved in the glycolysis, lactate, and Gln metabolic pathways. Gln tends to generate aspartate and is used by ME1 to generate pyruvate and NADPH for lipid synthesis. In addition, glutamate can be converted into isocitrate in cancer cells by isocitrate dehydrogenase (IDH1) under hypoxic conditions, which provides a non-canonical pathway for FA synthesis.[34] Lactate, in contrast, can be transported between different PDAC cells by MCT1 and MCT4. Furthermore, acetate can be synthesized de novo from acetaldehyde and converted into acetyl-CoA for lipid synthesis.[35]
The key enzymes in the network linking the metabolism of glucose, amino acids, and FAs in PDAC are shown in Figure 1. The causal relationships between these metabolic pathways increase the complexity and difficulty presented by heterogeneity.
Chemoresistance by Metabolic Reprograming in PDAC Cells Metabolic reprograming can activate important signaling pathways for PDAC chemoresistance Modulated protein kinase b/mechanistic target of rapamycin (AKT/mTOR) and/or adenosine-monophosphate activated-protein kinase (AMPK) /mTOR pathways, autophagy, and apoptotic pathwaysPDAC cells exhibit a specific metabolic pattern in the presence of a significant Warburg effect.[36] By blocking the TCA cycle, PDAC tumors tend to take up large amounts of glucose and accumulate lactate which is quickly pumped out of tumor cells, forming a low-energy condition. This starvation condition promotes continuous aberrant activation of survival signaling pathwayssuch as the AKT/mTOR and AMPK/mTOR signaling pathways.[37] Activated mTOR pathways can inhibit autophagy, while the negative regulation of mTOR (AMPK and p53 signaling pathways) promotes autophagy.[38] Studies have found that PDAC cells fail to survive in stressful environments, whereas the autophagy activator involved in AMPK/mTOR and AKT/mTOR pathways may promote PDAC cell growth.[37] Another study showed that the PI3K/AKT/mTOR pathway could induce drug resistance in PC via autophagy.[39]
Autophagy has been confirmed to be an important component of chemoresistance in several cancers. The major feature of autophagy is the formation of a phagophore. Specific proteins, including autophagy-related proteins (ATGs), form the autophagosome structure. Proteins involved in autophagy, such as the autophagy cargo-binding protein p62, have been proven to induce chemoresistance.[40] Several signaling pathways are involved in this process, such as arsenic trioxide-induced EGFR degradation, which is related to p62, which in turn enhances degradation, further improving secondary mutations of EGFR. Another example of ATG functions in chemoresistance is the promotion of endoplasmic reticulum (ER) stress by ATG3 in lung cancer cells. Inhibition of ATG3 translation results in the re-sensitization of cells resistant to erlotinib.[41]
Apoptosis is another important aspect of chemoresistance in PDAC. The main therapeutic mechanism of many first-line chemotherapeutic drugs is DNA damage, which activates the apoptotic pathway and causes cell death in tumor cells. In addition, activation of the AKT signaling pathway inhibits Bad by direct phosphorylation, thereby blocking Bim expression by phosphorylating and inhibiting members of the Forkhead family of transcription factors (FoxO), resulting in apoptosis blockade.[42] Blocking apoptosis further helps tumor cells survive chemotherapy. Thus, metabolic reprograming can also affect the apoptotic process to induce chemoresistance via the AKT signaling pathway. The signaling pathways that play key roles in autophagy and apoptosis are changed by metabolic reprograming, contributing to chemoresistance.
Lactate, the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, and histone hyperacetylationLactate has several key functions in PDAC, including energy transportation, microenvironment acidification, carcinoma-associated fibroblast (CAF) formation, and epithelial-mesenchymal transition (EMT). In addition to microenvironment changes, lactate plays an important role in metabolism-mediated signaling pathway reprograming and chemoresistance. Recently, lactate was reported to enter endothelial cells through a MCT-1 that is highly expressed in PDAC to trigger the phosphorylation/degradation of IκBα and stimulate an autocrine NF-κB/IL-8 pathway that drives cell migration and tube formation.[43] The NF-κB pathway not only promotes the proliferation and invasion of PDAC but also induces an anti-apoptotic effect and angiogenesis, which show a significant effect on the chemoresistance of PDAC.[44] High lactate levels caused by metabolic reprograming could affect the NF-κB pathway to promote chemoresistance in PC.
Furthermore, epigenetic research has revealed that lactate inhibits histone deacetylase activity and promotes changes in gene expression.[45] One study found that lactate can induce histone hyperacetylation corresponding to decreased chromatin compactness and enhance DNA double-strand break repair in tumor cells following neocarzinostatin, doxorubicin, or cisplatin treatment.[46] These results suggest lactate has a close relationship with chemotherapy resistance mediated by affecting signaling pathways and epigenetic characteristics.
Glutamine, chemoresistance, and the EGFR and β-catenin pathwaysAs mentioned before, the Gln metabolic pathway is reprogramed by oncogenic KRAS.[22] Gln is not only an important energy source in PDAC cells but also an important molecule that provides a nitrogen source for glycan biosynthesis through HBPs. One novel study found that gemcitabine sensitivity was restored in drug-resistant PDAC cells following treatment with a Gln analog, while a depression in glycan biosynthesis and downregulation of the EGFR signaling pathway were found after blocking the Gln metabolic pathway.[47] As most EGFR domain-containing proteins have multiple N- and/or O-linked glycosylations; we can infer that Gln-mediated HBP is a key step in EGFR expression. Further research revealed that Gln plays a significant role in maintaining the stemness of cancer cells via a redox-mediated mechanism mediated by β-catenin.[48]
In PDAC tumors, Gln is overexpressed to meet proliferation requirements and activate the EGFR pathway. Several years ago, the combination of gemcitabine and the tyrosine kinase inhibitor erlotinib was the first combination therapy to demonstrate survival benefits in PC.[49] Thus, Gln may lead to chemoresistance by affecting glycan biosynthesis and maintaining cancer cell stemness.
FASN and chemoresistanceFASN has been linked to intrinsic gemcitabine and radiotherapy resistance in PDAC, although the precise mechanism of FASN-induced gemcitabine resistance remains to be elucidated.[50] Subtle evidence has linked FASN and chemoresistance. First, it upregulates pyruvate kinase M2 (PKM2) expression, which modifies pyruvate synthesis, increasing the glucose consumption rate in PC cells, which promotes the Warburg effect and contributes to chemoresistance.[51] Second, recent research has shown a FASN inhibitor to decrease stemness in PC cell lines and maintain ER stress to re-sensitize those that are gemcitabine-resistant.[52] ER stress has been proven to play a key role in cancer cell death by activating the unfolded protein response. In chemoresistant PDAC cells, a high level of FASN may help cancer cells survive chemotherapy by stabilizing ER stress, and stable ER stress can, in turn, induce EMT in cancer cells to further develop resistance.[53] More studies are needed to understand the mechanisms connecting FASN, ER stress, and PDAC chemoresistance. Additionally, FASN is associated with cisplatin resistance by inducing apoptosis and decreasing drug efficacy, and its inhibition increases the sensitivity of cancer cells to chemotherapy.[54] However, more precise studies on FASN-mediated signaling pathway changes in PDAC are needed.
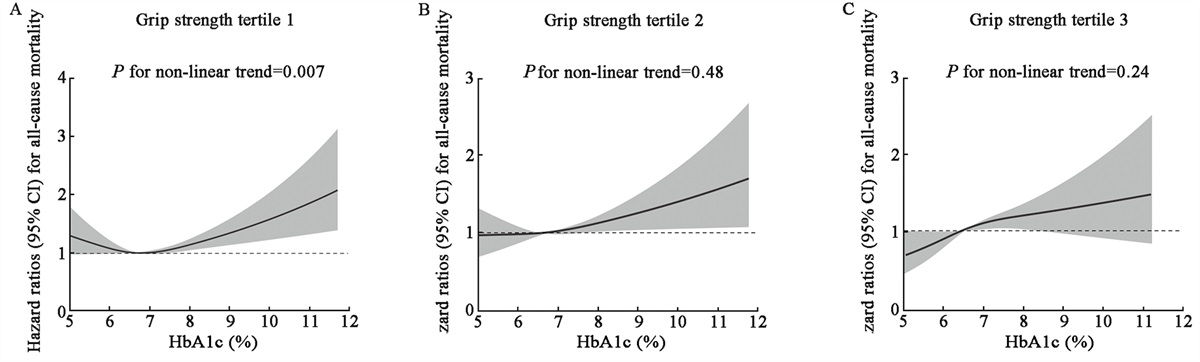
The role played by metabolic reprograming in modulating the signaling pathways that lead to chemotherapy resistance is illustrated in Figure 2.
 Figure 2:
Figure 2: Metabolic reprograming leads to signaling pathway remodeling that promotes chemoresistance. (A) Starvation condition in PDAC cells results in high oxygen consumption and an environment lacking energy, which activates the AMPK/mTORC1 pathway and AKT pathway to activate autophagy and prolong survival. (B) AKT can inhibit the downstream mediators FoxO1 and Bim to inhibit apoptosis, which is important in eliminating cancer cells. (C) High lactate levels can activate the NF-κB signaling pathway, which is closely related to chemoresistance. (D) Glutamine can promote glycosylation, which supports EGFR and further activates β-catenin to maintain the stemness of PC cells for chemoresistance promotion. (E) FASN also influences the ER stress response to promote survival and further chemoresistance. ADP: Adenosine diphosphate; AMP: Adenosine monophosphate; AMPK: AMP-activated protein kinase; AKT: Protein Kinase B; ATP: Adenosine triphosphate; EGFR:Epidermal growth factor receptor; EMT: Epithelial-mesenchymal transition; ER: Endoplasmic reticulum; FASN: Fatty acid synthase; FoxO1:Recombinant forkhead box protein O1; GFAT1: Glucosamine-fructose-6-phosphate aminotransferase 1; LDH: Lactate dehydrogenase; MCT1: Monocarboxylate transporter; MCT4: Monocarboxylate transporter 4; mTORC1: Mechanistic target of rapamycin complex 1; NADPH: Nicotinamide adenine dinucleotide phosphate; NF-kB: Nuclear factor kappa-lightchain-enhancer of activated B cells; ox PPP: Oxidated pentose phosphate pathway; PC: Pancreatic cancer; PDAC: Pancreatic ductal adenocarcinoma; PPP: Pentose phosphate pathway; UDP-G1cNac: Uridine diphosphate N-acetylglucosamine.
Ferroptosis and chemoresistanceThe field of ferroptosis research is increasing exponentially. This unique mode of cell death is driven by iron-dependent phospholipid peroxidation and regulated by cellular metabolic pathways, including redox homeostasis; iron treatment; mitochondrial activity; amino acid, lipid, and sugar metabolism; and various disease-related signaling pathways.
Cysteine, glutathione, and other molecules provide lipid antioxidant functions in PC, thus helping PC fight ferroptosis. Glutamate oxaloacetate transaminase 1 (GOT1), which is upregulated in PDAC, plays an important role in this process.[55]
GLUT1-mediated glucose uptake promotes glycolysis, promoting pyruvate oxidation, the TCA cycle, fatty acid synthesis, and lipid peroxidation-dependent ferroptosis. Studies have shown PDH kinase 4 to inhibit ferroptosis by blocking PDH-dependent pyruvate oxidation in PDAC.[56] Targeting metabolic weaknesses may induce ferroptosis in gemcitabine-resistant PC cells. Dihydroartemisinin (DHA) is a safe and promising therapeutic agent that induces ferroptosis preferentially in cancer cells. DHA was found to enhance the cytotoxicity of cisplatin and improve its effect on drug-resistant PC.[57] These findings indicate a novel target for gemcitabine-resistant PDAC.
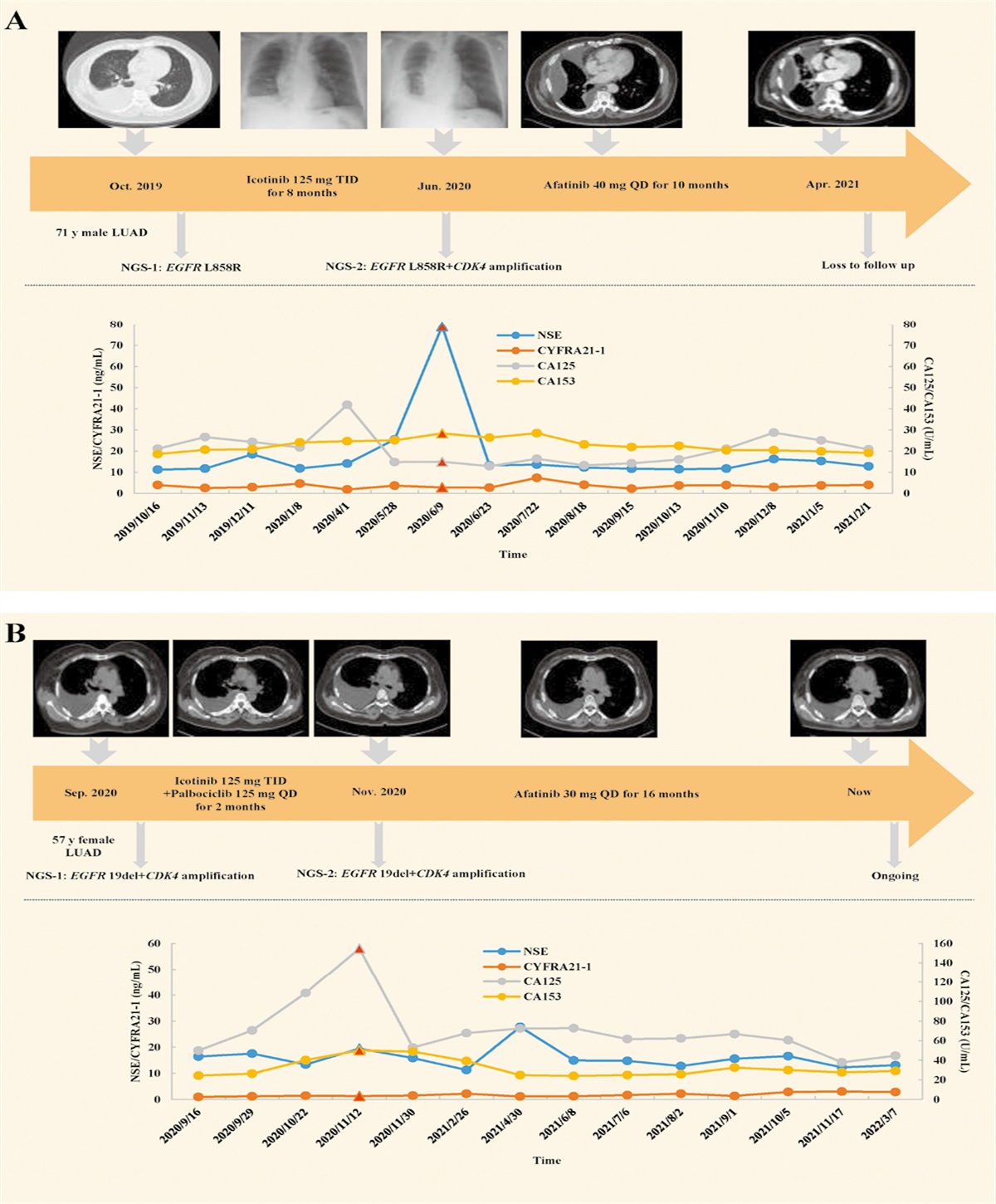
Metabolism-mediated transformation of the microenvironmentIncreasing evidence has shown that metabolic reprograming does not involve simple intercellular communication, instead substantially impacting the tumor microenvironment, which creates feedback affecting oncogenesis and chemoresistance. Extrinsic metabolic factors such as lactate primarily involve processes in the tumor microenvironment, including the acidification of the tumor microenvironment, stimulation of CAFs, and progression of EMT. CAFs and EMT are associated with chemotherapy resistance in PC,[58] and metabolic reprograming can also affect the immune system, thus creating a local immune environment favoring cancer cells and leading to EMT or other chemotherapy resistance-associated changes in PDAC.[59] Therefore, metabolic reprograming can induce chemoresistance by changing the microenvironment [Figure 3].
 Figure 3:
Figure 3: Metabolic reprograming changes the tumor microenvironment to promote chemoresistance. (A) CAFs tend to produce high levels of lactate that can be used by PDAC cells to maintain a high level of ROS to create a pseudohypoxic environment, which in turn supports CAFs. (B) Accumulation of lactate in the microenvironment increases the level of H+, which can promote EMT to induce chemoresistance. (C) Remodeling of immune cells can also promote EMT, and an increase in the TAM frequency can promote immunosuppression to support tumor escape. CAFs: Carcinoma-associated fibroblasts; EMT: Epithelial-mesenchymal transition; MCT1: Monocarboxylate transporter; MDSCs: Myeloid-derived suppressive cells; NK: Natural killer; OXPHOS: Oxidative phosphorylation; PDAC: Pancreatic ductal adenocarcinoma; ROS: Reactive oxygen species; TAMs: Tumor-associated macrophages; Treg: Regulatory T cells.
Secreted metabolic factors and CAF formationCAFs are the major cellular stromal components of the tumor microenvironment in most solid tumors, regulating the survival and proliferation of PDAC cells and inducing EMT. Recent studies have shown that CAFs consume more glucose and secrete more lactate than normal fibroblasts, feeding tumor cells rather than biosynthesis. In addition, cancer cell metabolic reprograming can help stabilize CAFs by elevating ROS production to promote glycolysis and create pseudohypoxic conditions for CAFs.[58] This bidirectional crosstalk proves that metabolic reprograming in PDAC promotes CAF proliferation, which induces chemoresistance.[60]
CAFs can increase extracellular matrix (ECM) production, trap cytokines in a fortified niche, and promote PDAC cell proliferation and EMT.[61] In a novel study, CAFs were shown to increase the release of exosomes when exposed to gemcitabine, which increased cell proliferation and survival in recipient epithelial cancer cells. Further mechanistic analysis showed that the expression of Snail, along with its target microRNA-146a, was increased in the exosomes secreted by CAFs. As Snail is an important molecule in the induction of pathological EMT in cancer cells, CAFs have been proven to play an important role in promoting EMT. The ECM forms a barrier to chemotherapeutic drug delivery. Overall, metabolic reprograming presents a substantial crosstalk that promotes CAFs in the PDAC microenvironment. CAFs are also linked to EMT via the remodeling of several mechanical signaling pathways, promoting changes that establish a chemotherapy-resistant microenvironment.
Metabolism-mediated regulation of immune cells and EMTImmune cells, including tumor-associated macrophages (TAMs), T lymphocytes, T regulatory cells (Tregs), natural killer (NK) cells, and myeloid-derived suppressive cells (MDSCs), are important microenvironment components that block the CD8+ T cell functions of tumor recognition and clearance. Extensive research has shown the PDAC microenvironment to be highly immunosuppressive. Crosstalk among different types of immune cells occurs to inhibit tumor growth, but metabolic reprograming disrupts normal metabolism in these cells leading to various functions. Under the severe energy competition pressure of the tumor microenvironment, T cells tend to become quiescent and long-living memory cells,[62] whereas Tregs and M2-like macrophages, which have low rates of glucose uptake and use lipids as substrates for OXPHOS, proliferate in progressing tumors as they can survive in low-glucose environments.[63]
Secreted lactate plays a key role in immune remodeling. First, the direct blocking of lactate export in T cells can induce T cell dysfunction, leading to immune remodeling.[64] Second, lactate can dampen NK cell functions, enhance MDSC functions, and induce M2-like TAM polarization.[65] Notably, M2-like macrophages can secrete a number of cytokines, such as interleukin (IL)-4, IL10, and IL-13, as well as glucocorticoids, which induce an alternative activation program of M2-like macrophages.[66] Further differentiation of M2-like macrophages can promote anti-inflammatory and immunosuppressive functions, leading to angiogenesis and tumor progression. Finally, lactate can induce an inflammatory environment to recruit immune cells, which accelerates the consumption of glucose and oxygen, induces the suppression of effector T cells and NK cells, and increases the inhibition mediated by MDSCs, Tregs, and M2-like macrophages.
Programmed cell death protein and its ligand (PD-1/PD-L1) is a popular target in immune therapy; however, PDAC shows resistance to PD-L1 inhibitors. Recent research showed that the high expression of GLUT-1 was correlated with tumor grade and density of PD-1 T cells, indicating their accumulation in glycolytic tumors. In a preclinical model, PD-1 CD8 tumor-infiltrating lymphocytes differentially infiltrated PDAC tumors obtained from cell lines with different metabolic consumptions, and tumors with metabolic reconnection achieved by knocking down the phosphofructokinase gene showed reduced PD-1 cell infiltration.[67] This research indicated that combining with metabolic inhibitors such as GLUT-1 inhibitors may make PDAC more sensitive to PD-1/PD-L1 inhibitors.
The most important function of chemoresistance involving changes in immune cells relies on the promotion of EMT. A novel study showed TAMs and M2-like macrophages to increase the chemotherapeutic resistance of PDAC tumors through EMT induction.[59] One hypothesized mechanism is that M2-like macrophages secrete the cytokines IL-1, IL-6, and tumor growth factor-beta (TGF-β), which play significant roles in triggering EMT progression in PDAC.[66] An emerging study also revealed that IL-4-induced M2-like macrophages could release deoxycytidine and induce gemcitabine resistance in PDAC, whereas this phenomenon was not observed in Lipopolysaccharide (LPS)-induced M1 macrophages. Compared with M1-type macrophages, M2-type macrophages have a greatly enhanced ability to exploit glycolysis for biosynthesis. Further inhibition of glycolysis or the PPP pathway showed that these treatments inhibited the ability of TAMs to produce deoxycytidine and modulated the gemcitabine sensitivity of PDAC cells.[68]
Intracellular signaling pathways and remodeling of the tumor microenvironment are highly linked to metabolism and chemoresistance. As the metabolic reprograming that occurs combines several factors of chemoresistance in a web-like pattern and maybe a weak point of tumors, numerous novel drugs have been designed based on highlighted metabolic targets for therapeutic purposes or for reversing chemoresistance.
Novel Therapeutic Drugs Based on Metabolic ReprogramingBecause gemcitabine-based treatment remains the first-line therapy for PDAC after decades of use, chemotherapy resistance remains a difficult problem for physicians. As new methods are sought, metabolic reprograming has been gradually accepted as a key step in PDAC oncogenesis, and targeting metabolism for novel treatments and for the reversion of chemoresistance is an emerging field of research. Those in the laboratory research and/or preclinical stages are reviewed in Table 1, and key metabolic targets and drugs are presented in Figure 4.
Table 1 - Clinical trials targeting metabolic reprograming in PDAC. Target Agent and method Stage NCT number Treatment setting Lysosome HCQ Phase II NCT01273805 Metastatic PC HCQ + GEM Phase I/II NCT01128296 Stage IIb or III PC HCQ + GEM/nab-paclitaxel Phase II NCT01978184 Resectable PC, Neoadjuvant HCQ + capecitabine and proton therapy Phase II NCT01494155 Resectable PC HCQ + Trametinib Phase I NCT03825289 Pyruvate Ddehydrogenase CPI-613 NCT01839981 Advanced or metastatic PC CPI-613 + mFOLFIRINOX Phase II NCT03699319 Locally advanced PC CPI-613 + FOLFIRINOX, CPI-613 + mFOLFIRINOX Phase III NCT03504423 Met
Comments (0)