
Remember me
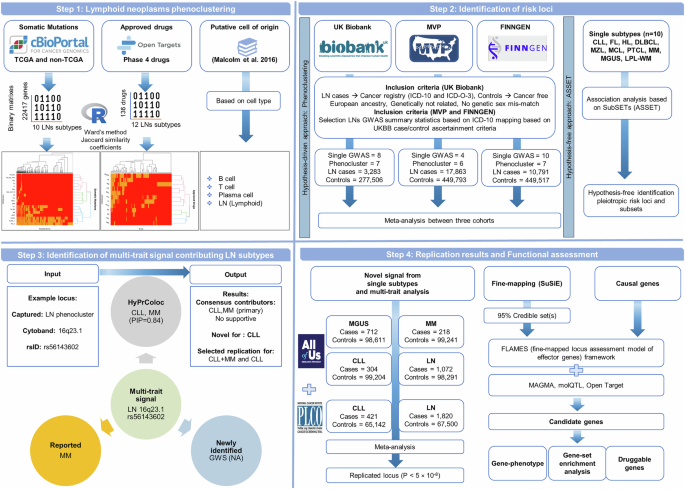
An overview of the study design is presented in Fig. 1, with detailed descriptions of each analytical step provided in the Supplementary Information. The complete computational pipeline, including scripts and workflows used for replication of all analyses and figures, is publicly available at https://github.com/biomguler/LN_Phenocluster/.
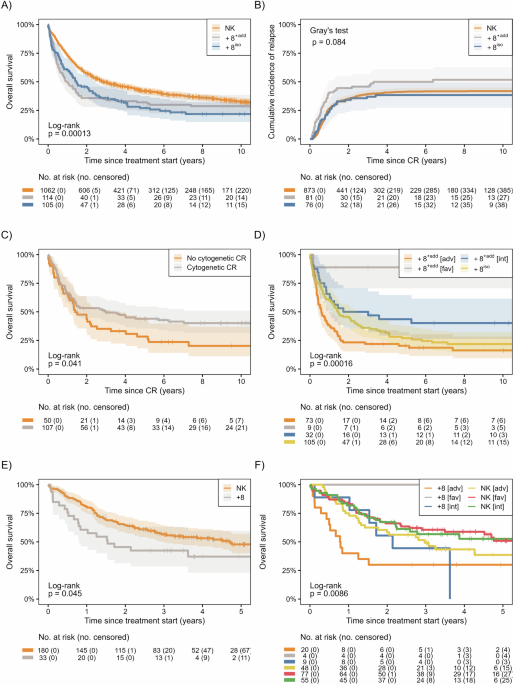
Fig. 1: Study design and graphical summary of the methods.
A comprehensive pipeline for identifying and characterizing multi-trait genetic signals associated with LN. The pipeline encompasses four organized steps: (1) LN phenoclustering, integrating somatic mutation data (cBioPortal; TCGA and non-TCGA datasets), drug information (Open Targets), and putative cell-of-origin annotations based on cell types, employing Ward’s clustering method to define subtype relationships; (2) Identification of genetic risk loci leveraging large biobank cohorts (UKB, MVP, and FINNGEN), through both hypothesis-free association analysis (SubSETs/ASSET approach) and hypothesis-driven phenocluster-informed analysis for identification of subtype-specific and pleiotropic loci across LN subtypes; (3) Characterization of multi-trait signals, illustrated with an example locus (LN phenocluster, 16q23.1, rs56143602), involving multi-trait colocalization (HyPrColoc), reported and newly identified association for subtypes, and identification of consensus contributor subtypes (CLL, MM primary); and (4) Replication and functional assessment, incorporating independent cohort meta-analysis (PLCO and AoU), fine-mapping (SuSiE), candidate gene prioritization using FLAMES (Fine-mapped Locus Assessment Model of Effector genes), functional annotation (FUMA: MAGMA, molQTL, Open Target), and detailed gene-based enrichment analyses to reveal LN biology and druggable genes.
Construction of LN phenoclusters using hierarchical clusteringTo group LN subtypes based on shared biological and clinical features, we performed hierarchical clustering using three modalities:
1.Cell-of-origin data were curated from the published literature [15,16,17,18], assigning each LN subtype to a major developmental lineage (i.e., B cell, plasma cell, and T cell).
2.Somatic mutation profiles were obtained from the cBioPortal database and converted into a binary matrix representing the presence or absence of mutations in 22,417 genes for each subtype (Supplementary Data 1).
3.Drug usage profiles were derived from the Open Targets Platform and transformed into binary matrices to indicate whether a given drug was approved for each LN subtype (Supplementary Data 2).
Each clustering was performed using Ward’s minimum variance method and Jaccard similarity coefficient, appropriate for binary data. Phenocluster definitions were guided by both algorithmic structure and biological interpretability (Supplementary Figs. 1–4).
An additional phenocluster including all LN subtypes was created to account for their shared hematopoietic origin.
Study populations and association testingWe conducted GWASs for individual LN subtypes and derived phenoclusters using three large, population-based cohorts: the UK Biobank (UKB), the Million Veteran Program (MVP), and FinnGen. Cohort description, data acquisition, and selection of cases and controls are extensively described in the Supplementary Information and Supplementary Tables 1–3. Summary statistics from each cohort were meta-analyzed using inverse-variance weighted fixed-effects models implemented in METAL [19]. We focused on only European ancestry due to a lack of statistical power for other ancestries.
Phenotype selection and association testingWe selected eight individual LN subtypes and seven phenoclusters for association analysis, retaining only phenotypes with ≥100 cases to minimize bias from case imbalance. In UKB, association testing was performed using REGENIE v3.2 [20], adjusting for age (at diagnosis for cases; at recruitment for controls), sex, genotyping array, and the first ten principal components (UKB Data-Field 22009) [21].
For FinnGen and MVP, phenocluster-level summary statistics were constructed by meta-analyzing available subtype-level results. Exceptions included the broad LN phenotype and MM-MGUS phenocluster, for which full summary statistics were directly available (Supplementary Table 4).
Association analysis based on subsets (ASSET)To complement the phenocluster-based approach in a hypothesis-free manner, we employed ASSET [14]. ASSET is a subset-based meta-analysis framework that systematically evaluates all possible combinations of traits to detect association signals, accounting for heterogeneity in genetic effects.
We performed both one-sided and two-sided ASSET analyses across ten LN subtypes: CLL, DLBCL, FL, HL, MGUS, MM, MCL, MZL, PTC, and LPL-WM. Subtypes were included if individual GWAS summary statistics were available from at least one cohort, with MCL and PTCL included based on data from FinnGen only.
One-sided ASSET was used to identify subsets of subtypes that showed associations in the same direction, either risk-increasing or risk-decreasing. Two-sided ASSET allowed for directional heterogeneity, enabling the detection of loci with opposite effects across subtypes by combining association signals using a chi-squared test.
Testing global genetic correlationTo quantify the shared genetic architecture among LN subtypes and phenoclusters, we estimated genome-wide genetic correlations using linkage disequilibrium score regression (LDSC), implemented with the LDSC v1.0.1 software [22]. Summary statistics from genome-wide association analyses of individual LN subtypes and phenoclusters were processed using the munge_sumstats.py utility provided in the LDSC package. Analyses were restricted to HapMap3 variants, following recommended best practices to ensure reliability of heritability and correlation estimates.
Variants with a minor allele frequency (MAF) below 5% were excluded from the analysis. In addition, we removed variants located within the extended major histocompatibility complex (MHC) region on chromosome 6 (25–35 Mb), due to the complex linkage disequilibrium patterns that can bias correlation estimates in this region. Bivariate genetic correlations were calculated between each pair of traits, and statistical significance was determined using a Bonferroni-corrected threshold of P ≤ 0.005, accounting for ten unique subtypes tested.
Definition of independent loci and genomic regionsTo define independent genome-wide significant (GWS) loci, we applied the clumping procedure implemented in PLINK [23] using a P-value threshold of 5 × 10−8, an R² threshold of 0.01, and a physical distance window of 1 megabase (Mb) around the index variant (command: -- clump -p1 5e-8 --clump-p2 5e-8 --clump-r2 0.01 --clump-kb 10000) and merged those loci with lead SNPs within 1 Mb of each other to obtain the final independently significant loci. For analyses involving individual LN subtypes, novel loci were defined as those not previously reported for the same subtype. Specifically, a locus was considered novel if its lead single-nucleotide polymorphism (SNP) was located outside a ±1 Mb window from any known lead variant and exhibited low LD (pairwise R² < 0.01) with previously reported associations, as detailed in Supplementary Table 5.
Identification of driver-subtypes and pleiotropic lociTo identify the specific LN subtypes contributing to multi-trait association signals and to classify pleiotropic loci, we used a three-step integrative strategy combining phenocluster-based and subset-based findings. First, we applied Hypothesis Prioritization in Multi-Trait Colocalization (HyPrColoc), a Bayesian framework that detects colocalized association signals and infers likely causal variants shared across traits. HyPrColoc groups traits based on shared regional association patterns and computes a posterior probability (PP) of colocalization for each cluster [24]. Analyses were conducted using default parameters, with prior.1 set to 1 × 10−4 and prior.c to 0.02, and with the branch-and-bound search algorithm enabled. Subtypes with regional PP values greater than 0.7 were designated as “primary contributors”, while those with lower support were labeled as “supportive contributors”.
Second, we examined subtype-specific GWAS results at each multi-trait locus. Subtypes were classified as primary contributors if they reached genome-wide significance (P < 5 × 10−8) and as supportive contributors if they showed suggestive significance (5 × 10−8 < P < 1 × 10−6). These annotations were based on results reported in Supplementary Table 6.
Third, we cross-referenced all identified loci with previously reported subtype-specific risk loci. If a subtype exhibited suggestive significance at a locus and had been previously implicated in association with a lead SNP located within ±500 kb and in LD (R² ≥ 0.01), it was also considered a primary contributor
By merging primary, supportive, and previously reported contributors, we generated a final list of associated subtypes for each locus. Loci were categorized as pleiotropic if two or more primary subtypes were implicated, as non-pleiotropic if only one primary contributor was identified, and as “potentially pleiotropic” if no clear primary or supportive contributor could be assigned. This classification allowed us to dissect the subtype-specific vs shared genetic basis underlying multi-trait associations.
Replication of novel lociWe attempted replication of novel associations using summary statistics from two independent cohorts: the All of Us (AoU) Research Program [25] and the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial [26]. For individual LN subtypes, replication was limited to CLL, MM and MGUS, for which data were available with sufficient statistical power in AoU and/or PLCO. Supplementary Table 4 shows LN phenotype definitions and case-control numbers.
For loci identified through phenocluster- or ASSET-based analyses, we selected subtypes for replication based on their contributor status. If the novel signal involved one or two contributing subtypes, replication was performed using single-subtype data or a subtype-specific meta-analysis within the replication cohort. If a locus involved three or more contributing subtypes, or if no specific contributors could be confidently assigned, replication was conducted using a broad LN phenotype, defined in AoU and PLCO as a composite of all available LN subtypes.
Meta-analysis of discovery and replication data was performed using inverse-variance weighted fixed-effects models implemented in METAL [19].
A locus was considered replicated if the effect direction was concordant with the discovery analysis, the effect size was of similar magnitude, and the combined meta-analysis reached genome-wide significance.
Statistical fine-mappingTo identify putative causal variants within associated loci, we performed statistical fine-mapping using the SuSiE (Sum of Single Effects) method [27] with both individual subtypes and phenocluster-level GWAS results. For each GWS locus, we defined a ±500-kilobase (kb) region around the lead SNP as the input window. LD reference matrices were generated using genotype data from 337,491 unrelated British participants of European ancestry in the UKB [28], ensuring population-matched LD structure for accurate posterior inference.
We used the susieR package (version 0.12.35) in R with default parameters. We required 95% credible sets to achieve a posterior inclusion probability (PIP) coverage of at least 0.95, with a minimum pairwise LD threshold of R² ≥ 0.5 to ensure variant correlation within sets. Loci located within the extended MHC region were excluded from fine-mapping. Seven of the fine-mapped loci did not yield credible sets and were excluded from downstream interpretations.
Functional annotation of variantsTo explore the molecular mechanisms underlying the identified association signals, we performed functional annotation of fine-mapped variants using the Ensembl Variant Effect Predictor (VEP, version 113) [29]. This included annotation of variant consequences, predicted functional effects, and overlap with known regulatory elements.
We also integrated cis-molecular quantitative trait locus (molQTL) data to assess the regulatory activity of credible set variants. These included expression (eQTLs), splicing (sQTLs), protein (pQTLs), transcript usage (tuQTLs), and single-cell expression QTLs (sceQTLs), sourced from multiple large-scale databases including the eQTL Catalog [30], Open Targets Platform [31], eQTLGen Consortium [32], and the UKB Pharma Proteomics Project [33]. We focused on QTLs derived from hematopoietic tissues and whole blood.
Locus to gene mappingTo prioritize candidate effector genes at associated risk loci, we applied a multi-pronged locus-to-gene mapping strategy integrating statistical, functional, and regulatory evidence. First, we used FLAMES (Fine-mapped Locus Assessment Model of Effector Genes), a machine learning-based framework that aggregates diverse genomic annotations to predict the most likely effector gene per locus [34].
Second, we conducted gene-based association testing using MAGMA [35], as implemented in FUMA (version 1.5.2) [36]. MAGMA integrates GWAS summary statistics and gene location to compute a gene-level test statistic. Genome-wide significance for MAGMA analyses was defined at a Bonferroni-corrected threshold of P = 2.63 × 10−6, corresponding to 19,010 tested genes.
Third, we used the Open Targets Locus2Gene scoring framework [37] to identify the most likely gene(s) at each locus based on proximity, functional consequence, and regulatory evidence from fine-mapped variants.
Fourth, we integrated cis-molecular QTL annotations from the previously described molQTL databases. Genes were considered supported if they were significantly regulated by variants within the 95% credible set.
Each gene was given a score of 1 if it was prioritized by a given method and of 0 if not. Scores across methods were averaged to generate a composite prioritization score per gene. Genes with support from multiple independent lines of evidence were flagged as high-confidence candidates for functional follow-up.
Enrichment analysis identified genes and drug targetsTo investigate the biological relevance and translational potential of the prioritized genes, we conducted a series of enrichment analyses focusing on tissue specificity, functional pathways, and therapeutic targeting. We first performed tissue- and cell-type-specific enrichment analysis using the Web-based Cell-type Specific Enrichment Analysis (WebCSEA) tool [38]. This tool evaluates gene expression patterns across 1355 human tissues and cell types and provides both nominal and permutation-based P-values for enrichment. Analyses were performed separately for the full set of prioritized genes, as well as the subset derived exclusively from novel loci.
To explore functional protein–protein interactions (PPIs), we queried the STRING database (version 12) [39]. Enrichment for Gene Ontology (GO) biological processes was assessed using STRING’s built-in annotation framework. Terms were considered significantly enriched if they met a false discovery rate (FDR) threshold of <0.05, and a minimum of two genes in the enrichment set was required to prevent false enrichment signals.
To assess therapeutic relevance, we investigated drug–gene interactions (DGI) using the Drug–Gene Interaction Database (DGIdb) [40]. Identified gene–drug pairs were annotated with Anatomical Therapeutic Chemical (ATC) codes from DrugBank. We then tested for enrichment of ATC first- and second-level categories using Fisher’s exact test, with significance defined at FDR < 0.05 relative to the full set of ATC annotations in DrugBank.
In parallel, we queried Open Targets for known drug interactions involving our prioritized genes, focusing on agents with approved or investigational indications based on ChEMBL annotations [41]. For each gene–drug pair, we manually obtained data on clinical status and indication using DrugBank and ClinicalTrials.gov to determine relevance to LN. Genes located in the MHC region were excluded from all enrichment and interaction analyses.
Comments (0)