
Remember me
Response and progression criteria are necessary for any malignancy to standardize assessment of efficacy across trials. In multiple myeloma (MM), the International Myeloma Working Group (IMWG) established universally accepted criteria for response and progressive disease (PD) anchored on serum and urine paraprotein as primary biomarkers of disease burden [1].
The therapeutic landscape of newly diagnosed MM (NDMM) evolves rapidly. Near two-thirds of patients treated with quadruplet therapy (QUAD) containing a proteasome inhibitor, an immunomodulatory agent, an anti-CD38 monoclonal antibody (anti-CD38 mAb), and dexamethasone followed by autologous stem cell transplantation (ASCT) now achieve stringent complete response and therefore have no detectable serum or urine paraprotein [2,3,4]. In this setting, the assessment of measurable residual disease (MRD) became crucial to refine prognostication [2,3,4,5,6,7,8]. With serial MRD assessment, some patients will have a rise in MRD without meeting IMWG criteria for PD. There is no consensus on how this scenario should be handled clinically and limited knowledge of its prognostic implication. We aim to explore the implications of disease progression defined by MRD rise.
We utilized the dataset from the completed MASTER trial [8], a single arm, multi-center, phase 2, MRD response-adapted trial for patients with NDMM, with enrichment for patients with high-risk disease. In brief, participants received 4 cycles of induction therapy with daratumumab, carfilzomib, lenalidomide, and dexamethasone (Dara-KRd), followed by ASCT. After ASCT participants received 0, 4, or 8 cycles of Dara-KRd according to MRD-status. Patients had MRD assessment after induction, after ASCT, and after each 4-cycle block of Dara-KRd consolidation. Treatment ceased if 2 consecutive MRD results <10−5, at which point patients were monitored conventionally and with MRD assessment at least yearly. Participants not reaching the criteria for treatment cessation received lenalidomide maintenance until progression.
We additionally included patients from a prospectively curated institutional database. Those patients received induction therapy with daratumumab, bortezomib, lenalidomide and dexamethasone (Dara-VRd) for 4 cycles, ASCT, and the same parameters for post-ASCT consolidation, maintenance as utilized in MASTER [9], yet treatment cessation was left at the discretion of the treating physician. Patients had a similar pattern of disease monitoring, including MRD, as in MASTER. All patients provided written informed consent to participate in the MASTER clinical trial (NCT05231629) and in the institutional database.
We assessed MRD regardless of concomitant IMWG response category using next-generation sequencing (clonoSEQ®, Adaptive biotechnologies, Seattle, WA). We annotated MRD in a continuous numeric scale. We did not attempt to distinguish results with no residual clonogenic sequences detected and results with sequences detected <10−6 and both were considered negative at 10−6 threshold [10].
We studied the implications of MRD progression (MRD-P), arbitrarily defined as a quantitative increase in MRD burden by at least 1 log10 above the nadir. For patients with MRD < 10−6, we considered 1 log10 increment, and therefore MRD-P, when MRD reached ≥10−5. Treatment modification with MRD-P without IMWG-defined PD was at the discretion of the treating physician. For patients with MRD-P without concomitant IMWG-defined PD we describe cumulative incidence of PD.
To minimize the impact of the heterogenous management and to contextualize the clinical implications of MRD-P, we describe survival free of failure of second-line therapy (SFF2T), defined as the time from progression event (MRD-P or PD not preceded by MRD-P) until the occurrence of IMWG-defined PD or death on second-line therapy, regardless of whether the second line was initiated due to MRD-P or IMWG-defined PD. Patients without PD on second-line therapy were censored at the time of the last follow-up. We also describe overall survival (OS) from the time of the progression event (MRD-P or PD). Alive patients were censored at the time of the last follow-up. Time-to-event endpoints were displayed using the method of Kaplan and Meier. All statistical analysis was performed using SPSS (SPSS Statistics for Macintosh, Version 29.0.1.1 Armonk, NY: IBM Corp). We considered statistical significance for P < 0.05.
We included 216 patients, 113 (52%) were MASTER clinical trial participants, and 103 (48%) were from the institutional database. Median follow-up is 39.5 months (45.3 months for MASTER and 28.8 months for institutional database). The median age was 61 years, 53% had MM with 1 or more high-risk chromosome abnormalities [HRCA, gain/amp 1q, t(4;14), t(14:16), del(17p)] (Table 1).
Table 1 Characteristics of patients.We obtained 966 MRD datapoints, with median of 4 (IQR 4–5) sequential MRD assessments per patient both in MASTER and in the institutional database. Overall, 78% achieved MRD negativity at 10−5, 68% at the 10−6 threshold.
Nineteen patients had MRD-P and 30 PD not preceded by MRD-P. Patients with MRD-P or PD not preceded by MRD-P had similar disease characteristics, and often had 1 or more HRCA along with other indicators of high risk (Table 1).
The median time from onset of therapy to MRD-P was 24.4 months and to PD not preceded by MRD-P was 23.2 mo. Among the 19 patients with MRD-P, 12 (63%) were on observation at time of MRD-P, 1 was receiving single-agent lenalidomide, 5 were receiving a QUAD, and 1 had just completed ASCT (Supplementary Fig. 1). Seventeen (89%) had most recent MRD assessment <10−5 including 10 (53%) with MRD < 10−6 (Supplementary Fig. 2). After MRD-P, 10 (53%) patients changed therapy, 9 to an anti-CD38 mAb-containing regimen, 5 continued same therapy, 2 continued observation, and 2 rapidly evolved with IMWG-defined PD (Fig. 1). Median time from MRD-P to PD was 10.1 months (Fig. 1). Of the 30 patients with PD not preceded by MRD-P, 19 were on observation at time of PD, 7 were receiving single agent lenalidomide, 2 dual maintenance, and 2 had just received ASCT (Supplementary Fig. 1). Twenty-four (80%) had most recent MRD < 10−5 including 19 (63%) with MRD < 10−6.
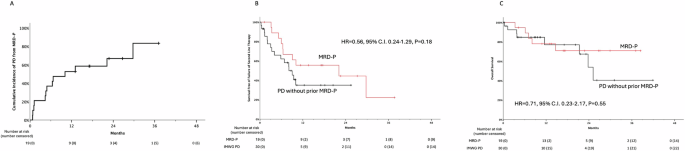
Fig. 1: Outcomes of patients with MRD-P.
Cumulative incidence of IMWG-defined PD among the patients with MRD-P (A). Survival free of failure of second-line therapy (SFF2T) of patients with MRD-P vs. those with PD not preceded by MRD-P (B). Overall survival of patients with MRD-P vs. those with PD not preceded by MRD-P (C).
Patients with MRD-P had 1-year SFF2T of 56% vs. 35% for patients with PD not preceded by MRD-P (HR = 0.56, 95% C.I. 0.24–1.29, P = 0.18). OS 2-year from progression event was 78% for MRD-P vs. 54% for PD not preceded by MRD-P (HR = 0.71, 95% C.I. 0.23–2.17, P = 0.55, Fig. 1).
The role of paraprotein as a pilar biomarker for the determination of PD is rooted in tradition and accepted by the scientific community and regulatory authorities when interpreting clinical trials for approval of new therapies. The present analysis, the first in the context of NDMM treated with QUAD + ASCT, demonstrates that a rise in MRD carries similar prognostic implications as an increase in paraprotein.
Most patients were on observation at the time of MRD-P and were started on combination therapy in reaction to MRD-P (Supplementary Fig. 1). In a setting where patients are on continuous therapy and treatment not modified by MRD-P (as in most clinical trials), the interval between MRD-P and PD would be even shorter than our observed 10.1 months. SFF2T and OS were similar between patients experiencing progression by MRD-P and those with progression by IMWG-defined PD. This analysis suffers from inevitable lead time and length time bias, yet those would skill the findings away from null, in favor of longer SFF2T in MRD-P group. In aggregate, our findings suggest that in the context of modern MM therapy, MRD rise is a reliable indicator of a thriving malignant plasma cell population that is unlikely to be controlled by previously utilized drugs.
Prior studies analyzed the implications of MRD resurgence. In the FORTE trial, among 70 patients who had received KRd +/- ASCT, achieved and subsequently lost MRD negativity, the median time from loss of MRD negativity to IMWG-defined PD was 22.3 months [11]. Patients in the GEM2012MENOS65/ GEM2014MAIN trials had received VRd + ASCT + dexamethasone, lenalidomide +/- Ixazomib maintenance. Among the 71 patients with MRD resurgence without concomitant PD, the median time for IMWG-defined PD was 39 months [12]. Unlike patients in FORTE and GEM2012MENOS65/ GEM2014MAIN, patients in our analysis were treated with anti-CD38 mAb, an intervention known to vastly improve the proportion of patients achieving MRD negativity and PFS [2,3,4]. Although the duration of follow-up differs, loss of MRD negativity appears understandably more common in these studies (23% and 27% respectively), than in ours (8%). Unlike ours, both these studies had more MRD resurgence events than IMWG-defined PD not preceded by MRD-P. In aggregate, these observations suggest that more indolent disease is responsible for slow rise and MRD resurgence with long intervals until PD with triplet therapy is often kept in MRD negative state after QUADs. Conversely, the cases with rising MRD despite QUAD and ASCT are intrinsically more aggressive and resistant to therapy, accounting for the short interval until PD and the relatively poor outcome. Therefore, MRD rise post QUAD + ASCT carries a different meaning than after lesser therapy.
Our analysis has limitations. We had a small number of events (itself a testament of the efficacy of QUADs + ASCT) and no standardized management of MRD-P. Given the limited follow-up, we could not address the impact of the timing of MRD-P and it is possible that MRD-P occurring much later after initial therapy is less ominous. We believe our findings should be confirmed by analyzing trials with serial MRD assessment after QUAD+/- ASCT.
This study challenges the paradigm of paraprotein elevation and measurability as minimal parameters for the definition of progression and for the experimental deployment of agents with new mechanisms of action.
Comments (0)