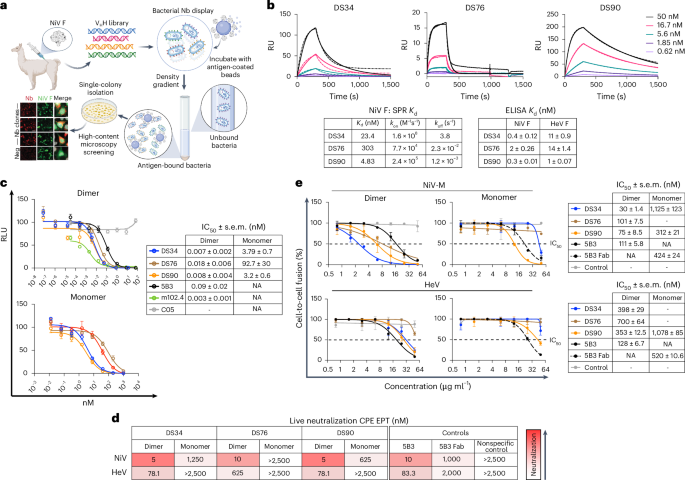
Structure-guided molecular glues
Targeted protein degradation is a proven paradigm for advancing the fields of drug discovery, biochemical regulatory networks, and proteostasis. Although several targeting strategies exist, molecular glues — small molecules that stabilize endogenous or non-native protein–protein interactions — are enticing because of their versatility, high cellular uptake and desirable pharmacological properties. Inducing protein degradation is a promising translational approach, particularly for targeting the classically ‘undruggable’ proteome. In RSC Chemical Biology, Ghosh et al. use X-ray crystal structures to inspire the de novo design of molecular glues with potential as future cancer therapies.
The authors determined the mechanism of action for the CDK12 inhibitor, SR-4835. The compound acts as a molecular glue that stabilizes interactions between CDK12 and its adaptor protein, DDB1, facilitating proteasomal degradation. By solving the DDB1–SR-4835–CDK12–cyclin K ternary complex structure, the authors leveraged knowledge of the protein–protein interaction interfaces to design molecular glues by converting established CDK12 inhibitors into potent cyclin K degraders. SR-4835 was found to occupy the binding pocket in CDK12 and bridged the protein complex through its benzimidazole ring; this benzimidazole moiety was appended onto the CDK12 inhibitor dinaciclib, which, in turn, induced degradation of cyclin K. The authors applied this approach to other purine scaffolds, finding that they degraded their target with varying potency. Interestingly, functional analysis revealed that while these glues induced degradation through complex formation, they did not considerably affect enzymatic activity. In vivo proteome-wide degradation analysis confirmed that the compounds selectively degraded cyclin K, validating that their chemical design approach yields successful drug candidates in a biological context.

Comments (0)