
Remember me
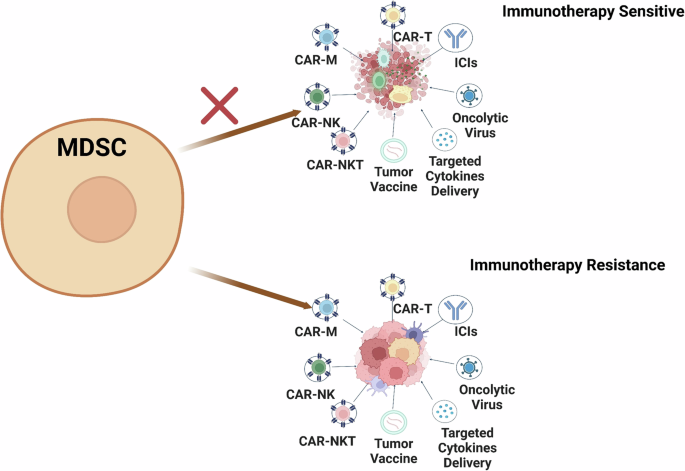




MDSC checkpoint blockade is at the forefront of modern immunotherapy. These checkpoints act as modulators of MDSC-driven immune suppression, offering promising avenues to enhance therapeutic efficacy against cancer [4]. These checkpoints consist of inhibitory or activating pathways and molecules predominantly expressed by MDSCs, which govern their immunosuppressive behavior.
Inflammatory cytokines and mediators, including TNF-α, IL-1β, PGE2, IL-6, and VEGF, TGF-β1, are among the tumor-associated factors that promote MDSC proliferation, expansion, and differentiation [31]. The balance between immunological activation and suppression can be significantly influenced by these checkpoints. Various checkpoints have been investigated in relation to MDSCs, but several key targets for reprogramming MDSCs include Cyclooxygenases, HIF-1α TNF-α, PD-L1, STAT3, C/EBPβ, c-Rel, PGE2, CCR2, PI3K, CHOP, TIPE2, CD300ld, FATP2, HIF-1α, Alox12/15, S100A8/A9, Dkk1, PLCγ 2, TRAIL-Rs, β2AR, LILRB, SLFN4, PERK, ASAH2, Tet2, CD45 phosphatase, RORC1, VISTA, SIRT 1, AMPK alpha 1, Retinoblastoma Gene 1, and Prokineticin 2 (BV8). These MDSC checkpoints orchestrate a highly efficient immunosuppressive network within tumors. Targeting these pathways offers a promising strategy to reprogram MDSCs, thereby reducing their immunosuppressive capacity and enhancing the efficacy of immunotherapies. By disrupting MDSC-mediated suppression, immune checkpoint inhibitors, CAR-T cells, CAR-Macrophages, and other adaptive therapies may achieve greater clinical success, especially in overcoming resistance in solid tumors. In Table 1, we summarized key MDSC-associated checkpoints, detailing the signaling pathways involved, mechanisms by which these checkpoints mediate immunosuppression, and potential drug candidates targeting these pathways. Some of these candidates are already FDA-approved for clinical use, while others are currently under investigation in clinical trials (Table 2).
Table 1 Summary of MDSC checkpoints and the potential drug candidate in pro-clinical settings.Table 2 Summary of clinical trials targeting MDSCs checkpoints as immunotherapies.In this review, we also categorize MDSC checkpoints into seven key areas: amino acid metabolism (CHOP, PERK); lipid metabolism (Alox12/15, FATP2, ASAH2, SLFN4, COX, PGE2); mitochondrial metabolism and bioenergetics (AMPKα1, PI3K, SIRT1); hypoxia and oxidative stress (HIF-1α, DKK1); epigenetic and transcriptional regulation (C/EBPβ, STAT3, c-Rel, Tet2, Retinoblastoma Gene 1); membrane receptor signaling (CD74, TRAIL-R, LILRB, PD-L1, VISTA, CD300ld, BV8 receptor); TLR/NF-κB and MAPK signaling (S100A8/A9, TIPE2, CD45 phosphatase, TNF-α, PLCγ2). This categorization highlights how MDSCs leverage diverse metabolic pathways and signaling mechanisms to promote immune suppression (Fig. 2). We propose that targeting these distinct metabolic checkpoints may offer novel therapeutic approaches for overcoming cancer resistance to immunotherapy.
Fig. 2: Key signaling pathways involve MDSC checkpoints.
MDSC checkpoints pair and bind with their ligands on the cell membrane (top), and this activates key signaling pathways leading to regulating MDSC expansion, survival, and immunosuppressive activity (top).
Targeting checkpoints involved in lipid metabolismFATP2The trafficking of lipids across cellular membranes is mediated by various proteins, fatty acid transport protein (FATP), CD206, and CD36. Among these, the FATP family, consisting of FATP1 through FATP6, functions both as acyl-CoA synthetases (ACS) and as long-chain FA transporters [32]. In cancer, polyunsaturated fatty acids (PUFAs) have been shown to disrupt normal myelopoiesis in the bone marrow of tumor-bearing mice, leading to an increased accumulation and enhanced activity of PMN-MDSCs via the JAK/STAT3 signaling pathway [33]. This PUFA-driven effect on MDSCs was significantly reduced when STAT3 phosphorylation was inhibited by the JAK inhibitor JSI-124. FATP2 is selectively upregulated in both murine and human G-MDSCs, driven by GM-CSF through STAT5 activation [34]. FATP2-mediated immunosuppression involves the assimilation of arachidonic acid, a precursor for PGE2 production. Deletion of FATP2 eliminates the suppressive function of PMN-MDSCs, slowing tumor progression without affecting CD8+ T cell activity or the function of other immunosuppressive cells such as tumor-associated macrophages and monocytic MDSCs [35]. In addition, signaling via the β2-adrenergic receptor enhances fatty acid oxidation (FAO) and increases the expression of the FA transporter CPT1A in MDSCs, further promoting their immunosuppressive function [36]. This signaling pathway activates the arachidonic acid cycle, increasing PGE2 release and driving further immunosuppression [37]. PGE2 produced by MDSCs can elevate PD-L1 expression in both intratumoral MDSCs and TAMs, contributing to the differentiation of IL-10-producing T cells into dysfunctional, and exhausted T cells [38].
Targeting the COX2/mPGES1/PGE2 signaling pathway by blocking FATP2 offers a promising strategy to mitigate PD-L1-mediated immune suppression. These evidences suggests that FATP2 is a pivotal regulator of MDSC function, making it a promising therapeutic target. By inhibiting FATP2, it is possible to reduce MDSC-mediated immune suppression, potentially enhancing the efficacy of cancer immunotherapy. Furthermore, combining FATP2 inhibition with checkpoint blockade therapies such as anti-PD1, as well as adoptive cell therapies like CAR-T, CAR-NK, and CAR-M therapies, could further enhance anti-tumor immune responses. Future research should aim to elucidate the molecular mechanisms underlying FATP2’s interaction with CD36 and FA-binding proteins in MDSC lipid uptake, as well as to evaluate the therapeutic efficacy and safety of targeting FATP2 in combination with other immunotherapies.
Alox12/15Arachidonate 12/15-lipoxygenase (ALOX12/15) is a key enzyme involved in the metabolism of polyunsaturated fatty acids (PUFAs), primarily arachidonic acid, which is oxidized to produce bioactive lipids such as 12-hydroxyeicosatetraenoic acid (12-HETE) and 15-hydroxyeicosatetraenoic acid (15-HETE). These metabolites, along with others like prostaglandin E2 (PGE2) and eicosanoids, play a pivotal role in the immunosuppressive functions of MDSCs within the tumor microenvironment [39]. Research by Tang et al. has shown that IL-13 induces MDSCs to overexpress 15-lipoxygenase (ALOX15), leading to increased production of lipid mediators that enhance MDSC immunosuppressive activity [40]. Specifically, eicosanoids produced by ALOX15, such as 15-HETE, have been found to promote MDSC recruitment, growth, and activation, thereby contributing to the establishment of an immunosuppressive tumor microenvironment [10]. These eicosanoids dampen immune responses by inhibiting effector T cells and NK cells, and promoting the expansion of Tregs, which facilitates tumor progression. Further investigations have highlighted that deletion of the ALOX12/15 gene in tumor-associated PMN-MDSCs alters their metabolome and gene expression profiles [41]. The deletion enhances immune responses by upregulating genes involved in complement activation, neutrophil-mediated immunity, monocyte chemotaxis, and antigen presentation. Moreover, inhibition of ferroptosis, a form of iron defendant-regulated cell death mediated by ALOX12/15, has been shown to augment the anti-cancer effects of immune checkpoint inhibitors and to suppress tumor growth [42]. This suggests that ALOX12/15 not only supports MDSC-mediated immunosuppression but also plays a critical role in ferroptosis.
Given the central role of ALOX12/15 in MDSC biology, targeting this enzyme offers significant therapeutic potential. Blocking ALOX12/15 could disrupt the synthesis of lipid mediators that decrease immunity, such as PGE2 and 15-HETEs, reducing the immunosuppressive capacity of MDSCs and enhancing the immune system’s ability to attack the tumor. By impairing MDSC function, ALOX12/15 inhibitors could also promote a pro-inflammatory tumor microenvironment, facilitating the homing and activation of cytotoxic immune cells, including CD8+ T cells and NK cells. Furthermore, targeting ALOX12/15 could synergize with other immunotherapeutic approaches, such as cancer vaccines, immune checkpoint blockade, and adoptive cellular therapies. Further research on ALOX12/15 inhibitors is crucial to understanding their therapeutic potential and their role in MDSCs ferroptosis, which could enhance anti-tumor immunity and cancer immunotherapy efficacy.
PGE2Prostaglandin E2 (PGE2) is a key mediator in the biology of MDSCs, playing a significant role in chronic inflammation and tumor progression. PGE2 facilitates MDSC expansion and enhances their suppressive impact on T cell-mediated immune responses [38]. It modulates the activity of enzymes associated with MDSC function, including iNOS, indoleamine 2,3-dioxygenase, and arginase 1, through binding to four distinct receptors: EP1 through EP4 [43]. Studies have demonstrated the pivotal role of the PGE2-COX2 signaling pathway in regulating MDSC immunobiology. Rodriguez et al. showed that PGE2-COX2 drives the expression of the immunosuppressive gene arginase 1 in MDSCs. Inhibiting COX-2 resulted in reduced arginase I expression and enhanced lymphocyte-mediated antitumor responses [43]. Additionally, COX2 activity in MDSCs is associated with the generation of mitochondrial ROS, and decreasing COX2 reduces ROS levels, compromising MDSCs’ inhibitory capacity [38, 39]. Moreover, PGE2 released by tumors upregulates CD73 expression in monocytic MDSCs, leading to increased suppression of T cell activity through elevated adenosine levels. Lowering adenosine levels with PEGylated adenosine deaminase has been shown to enhance CD8+ T cell activation and improve the response to immune checkpoint inhibitor therapy [44]. Furthermore, PGE2 secreted by MDSCs in ovarian cancer promotes the expression of PD-L1 and the emergence of cancer stem-like cells. It upregulates PD-L1 expression in ovarian cancer cells via the mTOR signaling pathway, contributing to tumor progression [45]. Targeting EP receptors, particularly EP2 and EP4, presents a promising approach for enhancing the effectiveness of adoptive immunotherapy. Clinical studies evaluating EP4 antagonists (such as E7046) and dual EP2/EP4 antagonists (such as TPST-1495) in advanced malignancies are underway. The COX-2-PGE2 signaling cascade plays a crucial role in modulating the tumor microenvironment (TME) by influencing immune cell polarization, activating cancer-associated fibroblasts, and promoting ECM remodeling, angiogenesis, and T cell exclusion. Its involvement in epithelial-to-mesenchymal transition (EMT) and crosstalk with pathways like Wnt/β-catenin and TGF-β highlights its importance in tumor progression. Understanding these mechanisms could help overcome resistance to COX-2 inhibitors and guide novel combination therapies.
COXCyclooxygenases (COX) are enzymes crucial to producing prostanoids, including prostaglandins, prostacyclins, and thromboxanes. There are two main isoforms: COX1, considered a “housekeeping” enzyme, and COX2, an inducible isoenzyme. COX2 is expressed at low levels in most tissues but can be highly expressed in response to various stimuli, including inflammatory mediators and tumor promoters [46]. Targeting the COX pathway in MDSCs has shown promise in enhancing immunotherapeutic efficacy. COX2 inhibitors reduce MDSC prevalence and impair their suppressive functions by decreasing ARG1 expression and inhibiting mitochondrial ROS production within MDSCs [47]. This highlights COX2 as a potential therapeutic target to mitigate MDSC-induced immunosuppression in the tumor microenvironment. Studies have explored the efficacy of targeting the end receptors of COX1/2 metabolites using selective EP4 receptor antagonists like MF-766 [48]. Furthermore, in mouse models, MF-766 enhances the efficacy of anti-PD-1 therapy by promoting the infiltration of CD8+ T cells, NK cells, and conventional dendritic cells into the tumor microenvironment, shifting macrophage phenotypes toward M1-like ones, and reducing the number of granulocytic MDSCs. Additionally, Li et al. found a correlation between COX-2 expression and key immunosuppressive genes in MDSCs in nasopharyngeal cancer (NPC). Inhibiting COX-2 attenuated the rise of COX-2 levels and epithelial-mesenchymal transition (EMT) scores in NPC cells driven by MDSCs, suggesting COX-2 as a critical checkpoint for MDSC–tumor cell interactions and tumor promotion [47]. In the same vein, Prima et al. reported a significant decrease in PD-L1 transcription after inhibiting PGE2 anabolism by COX2 inhibitors or enhancing PGE2 catabolism. This indicates the role of the COX2/mPGES1/PGE2 axis in controlling PD-L1 expression in myeloid cells within the tumor microenvironment [49]. Furthermore, Muthuswamy et al. demonstrated that COX2-specific inhibitor Celecoxib effectively counteracted the suppressive influence of myeloid cells on effector immune cells by inhibiting COX2, IDO, and IL-10 expression. In addition, Veltman et al. reported large numbers of infiltrating MDSCs were found to co-localize with COX-2 expression in areas of active tumor growth. Treatment with celecoxib was shown to effectively reduce prostaglandin E2 levels both in vitro and in vivo. In mesothelioma tumor-bearing mice, celecoxib administration prevented the local and systemic expansion of all MDSC subsets, leading to impaired MDSC function characterized by decreased levels of ROS and NO. Additionally, celecoxib treatment reversed T-cell tolerance, thereby enhancing the efficacy of immunotherapy [50]. Collectively, COX2 inhibitors show promise in diminishing the MDSC population and inhibiting their suppressive roles, positioning them as potential trajectories in cancer immunotherapy. However, further research is required to validate their safety and utility in combination with other immunotherapy.
ASAH2Acylsphingosine amidohydrolase 2 (ASAH2) is a neutral ceramidase that plays a crucial role in sphingolipid metabolism in MDSCs, helping them resist ferroptosis. This hydrolytic enzyme is encoded by the ASAH2 gene which is critical in ceramide metabolism [51]. Multiple groups reported upregulation of ASAH2 expression in both colon cancer cells and intratumoral MDSCs. They found that ASAH2 functions as a survival factor for MDSCs via the p53 pathway [52]. The researchers developed a small molecule inhibitor to block ASAH2 function. Inhibition of ASAH2 led to increased p53 protein stability, upregulation of Hmox1 expression, suppression of ROS generation, and MDSC death via iron-dependent lipid peroxidation. Additionally, the inhibitor treatment reduced glutathione and cysteine uptake in MDSCs, indicating that it targets glutathione synthesis from cysteine [53]. These findings highlight the role of ASAH2 as an important checkpoint and ferroptosis regulator in MDSCs. Coant and colleagues demonstrated that inhibition of ASAH2 triggers the dephosphorylation of GSK3β, necessary for the phosphorylation and degradation of β-catenin. They found that ASAH2 inhibition in colorectal cancer cells leads to decreased GSK3β phosphorylation and activation of AKT, a critical cell growth target. This inhibition also causes AKT dephosphorylation and dysfunction. The study suggests that ASAH2 controls the basal activation of AKT, a major pathway employed by MDSCs to drive immunosuppressive phenotypes, making it a potential checkpoint for colon cancer treatment. In a xenograft model, ASAH2 inhibition resulted in elevated ceramide levels, reduced proliferation, and delayed tumor growth, confirming the functional requirement of neutral ceramidase in colon cancer control and progression [52]. These results highlight the significance of ASAH2 in controlling ferroptosis in MDSCs and imply its possibility as a target to overcome MDSCs-mediated immunosuppression.
SLFN4Targeting Schlafen 4 (SLFN4) in MDSCs presents a promising strategy for overcoming immune suppression within the TME. SLFN4 is a member of the Schlafen family of proteins that plays a key regulatory role in the differentiation, recruitment, and activation of MDSCs [54]. Its expression is induced by type 1 interferons, particularly IFN-α, primarily produced by plasmacytoid dendritic cells (pDCs), and it has been shown to significantly enhance the immunosuppressive capacities of MDSCs [55]. In gastrointestinal malignancies, particularly during Helicobacter pylori infection, SLFN4 expression is upregulated and associated with the expansion of SLFN4+ MDSCs in the gastric TME. This process is mediated by the Sonic Hedgehog (SHH) signaling pathway, which promotes metaplastic transformation and enhances the immunosuppressive activity of MDSCs through mechanisms involving key suppressive factors such as Arg-1 and iNOS. SLFN4+ MDSCs also express microRNA miR130b, which mediates T-cell suppression, further contributing to the pro-tumorigenic environment [56]. The ability of SLFN4 to drive immune suppression by promoting MDSC polarization and activation suggests that it may play a similar role in other solid tumors. Indeed, SLFN4 knockdown experiments have shown that inhibition of this protein diminishes the immunosuppressive properties of MDSCs, reducing levels of Arg-1 and iNOS, and consequently restoring T cell function [57]. This suggests that targeting SLFN4 could impair MDSC function, alleviate immune suppression, and enhance the efficacy of other immunotherapeutic approaches. From a therapeutic standpoint, pharmacological inhibition of SLFN4 either through small molecules like sildenafil or genetic approaches has demonstrated efficacy in preclinical models. For instance, inhibiting SLFN4 in mouse models of H. pylori-induced gastric metaplasia not only attenuated MDSC-mediated immune suppression but also reduced the proliferation of cancer stem cells [58]. Targeting SLFN4 in MDSCs offers a dual benefit: it could reduce the immune suppression exerted by MDSCs, thereby lowering their inhibitory effects on T cells, while also modulating the TME to become less favorable for tumor growth. In this context, SLFN4 blockade could synergize with existing immunotherapies. For example, combining SLFN4 inhibition with immune checkpoint inhibitors (such as anti-PD-1 or anti-CTLA-4) could enhance T cell activity by not only relieving checkpoint-mediated inhibition but also reducing the immunosuppressive influence of MDSCs. Future research should explore the signaling pathways regulating SLFN4 expression and its effects on MDSC function, particularly STAT3 and NF-κB. Understanding SLFN4’s interactions could reveal how MDSCs maintain suppressive phenotypes and identify targetable metabolic vulnerabilities with SLFN4 blockade.
Targeting checkpoints involve in amino acid metabolismCHOPC/EBP homologous protein (CHOP), a crucial transcription factor, acts as a checkpoint for MDSCs in executing their immunosuppressive functions. Experimental evidence suggests that CHOP-deficient MDSCs can transition from immune suppressors to immune stimulators, acquiring some dendritic cell phenotypes and functionalities [59]. This functional polarization of MDSCs has been linked to factors such as endoplasmic reticulum (ER) stress, acidosis, and hypoxia, which significantly upregulate CHOP expression in intratumoral M-MDSCs and bone marrow-residing PMN-MDSCs [13]. The increase in CHOP expression is associated with peroxynitrite and ROS levels in the tumor microenvironment. Notably, CHOP is primarily expressed by myeloid cells infiltrating the tumor microenvironment, excluding macrophages [60]. Knocking out the Chop gene significantly inhibits tumor growth by altering MDSC activity, reducing their immunosuppressive functions, including diminished arginase-1 and ROS levels. Chop deficiency enhances IFN-γ-producing CD8+ T cell recruitment to tumors, reversing tumor growth when CD8+ T cells are reduced. MDSCs from Chop-deficient mice gain antigen-presenting capabilities, reflecting a functional shift. Additionally, CHOP deficiency reduces C/EBPβ, p-STAT3 activity, and IL-6 production, further impacting tumor progression [13]. Importantly, the restoration of tumor growth and MDSC suppressive activity in CHOP-deficient mice through IL-6 overexpression highlights CHOP’s regulatory role in MDSC behavior via IL-6 production. Notably, immunosuppressive activity is primarily observed in intratumoral and vascular MDSCs, whereas splenic MDSCs show minimal suppression of nonspecific T-cell proliferation. This distinction aligns with the specialized immunosuppressive role of MDSCs in the tumor microenvironment. Furthermore, lncRNAs play a critical role in modulating MDSC functions within the tumor, further emphasizing the complexity of their regulation [61]. A recently identified lncRNA, Lnc-CHOP, has been found to influence the immunosuppressive behavior of MDSCs. Lnc-CHOP promotes C/EBP activation and enhances the expression of immunosuppressive genes in MDSCs, whereas lnc-C/EBP suppresses C/EBP activation, leading to decreased immunosuppressive function and reduced MDSC differentiation [60]. Future research should explore CHOP’s role in MDSC-mediated immunosuppression during tumor progression, its interaction with cellular stress responses, immune checkpoint molecules, and tumor microenvironment. Understanding CHOP’s impact on T cell and NK cell functions is crucial. Identifying CHOP-related biomarkers could improve patient selection for CHOP-targeted therapies, making cancer immunotherapy more precise and effective.
PERKProtein Kinase R-like Endoplasmic Reticulum Kinase (PERK) is a type I integral transmembrane protein crucial for the cellular unfolded protein response triggered by ER stress resulting from the accumulation of unfolded or misfolded proteins. Several studies have highlighted the capability of targeting the PERK axis to reprogram MDSCs from immunosuppressive to antitumoral phenotype [62]. Research by Mahadevan et al. demonstrated that when tumor cells undergo ER stress, they can induce stress in intratumoral myeloid cells, resulting in upregulation of various tumor-promoting genes like Arg-1 and iNOS, accelerating tumor growth [63]. Additionally, recent findings suggest that the integrated stress response can drive PD-L1 overexpression in lung cancer [64]. Interestingly, Mohammed et al. reported elevated PERK signaling in tumor-derived MDSCs, and inhibition of this signaling reprogrammed MDSCs into effector antitumoral myeloid cells [62]. These reprogrammed MDSCs engulfed tumor cells, co-activated anti-tumor CD8+ T-cell function, and served as professional antigen-presenting cells. The disturbance of NRF2-driven antioxidant capability and mitochondrial respiratory equilibrium in PERK-deficient tumor-MDSCs impaired their ability to suppress CD8+ T cells. Moreover, cytosolic mitochondrial DNA elevation induced by reduced NRF2 signaling in PERK-deficient MDSCs led to STING-dependent production of anti-tumor Type-I Interferon. The immunoinhibitory capability of PERK-ablated MDSCs was restored by blocking Type-I interferon receptor-I, conditionally via deleting STING or reactivating NRF2 signaling. Furthermore, intravenous administration of MDSC-specific PERK knockout resulted in significant tumor growth suppression [62]. Studies also evaluated the synergistic effect of PERK inhibitors with anti-PD-L1 in the B16F10 tumor model, showing promising outcomes. PERK has been linked to MDSCs' “well-being” and prevents STING activation through its antioxidant properties. Additionally, attenuated tumor growth and activated anti-tumor T cell immunity were observed when PERK was deleted or pharmacologically inhibited in mice harboring melanoma [65]. Recently, Liu et al. found that HSPC reprogramming into committed MDSC precursors in the spleen was mediated via PERK-ATF4-C/EBPβ signaling [66]. In addition, pharmacological and genetic suppression of this pathway prevented the differentiation of myeloid descendant cells into MDSCs, leading to significant tumor regression in mice [67].
Targeting mitochondrial metabolism and bioenergeticsAMPK alpha 1AMP-activated protein kinase (AMPK) is a heterotrimeric serine/threonine kinase complex present in all animals, serving as a crucial metabolic sensor to maintain cellular energy balance during stress. This complex consists of catalytic subunits (AMPKα1 or AMPKα2) and regulatory subunits (AMPKβ and AMPKγ) [68]. Studies by Trillo Tinoco et al. revealed a significant association between AMPKα signaling and the immunosuppressive functions of MDSCs in tumors. They found heightened AMPKα activity in MDSCs from tumor-bearing mice and human ovarian cancer patients, driven by GM-CSF from cancer cells via STAT5-dependent transcription of the Ampkα1 gene. Inhibiting AMPKα slowed tumor growth, weakened MDSC suppression, activated antitumor CD8+ T-cell immunity, and enhanced the success of CAR T-cell therapy. Conversely, stimulating AMPKα signaling increased MDSC immunoregulatory functions [69]. Similarly, deleting AMPKα1 gene altered the differentiation pathway of M-MDSCs, leading them to adopt a cytotoxic role against tumors through nitric oxide synthase 2, highlighting the central role of AMPKα1 in MDSC-mediated immunosuppression. Various studies have demonstrated that AMPK activation inhibits the growth and negative functionality of MDSCs, suggesting it is a viable therapeutic target in immuno-oncology. Trikha et al. found that OSU-53 administration triggered AMPK phosphorylation, reducing nitric oxide production, MDSC migration, and IL-6 expression. Treatment with OSU53 decreased the immunosuppressive effects of murine MDSCs and increased T cell functionality. Moreover, AMPK is also essential for the differentiation of bone marrow cells into endothelial progenitor cells and mediates the differentiation of CD11b+/Gr-1+ MDSCs in tumor-bearing hosts. Inhibition of AMPK activity through Comp-C treatment reduced glucose uptake rates without impacting overall cell growth or viability [70]. Moreover, Adeshakin et al. demonstrated that metformin, an anti-diabetic drug, activates AMPK and enhances CHOP expression, thereby increasing stress responses in an anchorage-independent B16F10 melanoma model. This results in reduced antioxidant activity and the accumulation of misfolded proteins, sensitizing tumor cells to anoikis. This metabolic reprogramming could disrupt the supportive environment for MDSCs [71]. Consequently, targeting AMPKα1 in MDSCs, in combination with adaptive cellular therapies or immune checkpoint inhibitors (ICIs), could enhance the effectiveness of cancer treatments. However, care must be taken when targeting AMPKα1, as it plays a crucial role in maintaining energy balance across various cell types. Inhibiting AMPK in MDSCs may unintentionally disturb energy regulation in healthy tissues, potentially causing adverse side effects. Therefore, therapeutic strategies must carefully balance targeting AMPK in MDSCs while minimizing potential harm to non-cancerous cells.
PI3KPhosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) plays a pivotal role in regulating cell cycle processes, such as proliferation, quiescence, and energy metabolism. In the context of MDSCs, PI3K signaling is integral to several cellular functions, including cytokine production, homeostasis, glucose and lipid metabolism, proliferation, and recruitment [72]. These processes collectively drive the immunosuppressive capabilities of MDSCs. The PI3K/AKT/mTOR pathway is particularly important for the development and immunosuppressive behavior of MDSCs in both chronic inflammation and malignancies. The PI3K family includes PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ, with PI3Kδ and PI3Kγ being the primary isoforms implicated in promoting survival and immunosuppressive functions in myeloid cells [73]. This signaling pathway can shift cellular energy generation from glycolysis to oxidative phosphorylation (OXPHOS), which supports MDSC survival and their suppressive activity. PI3Kγ specific blockade to target these cells and improve tumor outcomes has been widely studied in recent years with propitious outcomes in the preclinical stage. Inhibiting the PI3K/AKT/mTOR circuit compromises the viability and function of MDSCs [74]. Dysregulation of PI3K/AKT signaling has been linked to the accumulation of MDSCs in multiple tissues, including the bone marrow, secondary lymphoid organs, sites of chronic inflammation, and tumors. Importantly, targeting specific isoforms of PI3K, such as δ and γ, using inhibitors like IPI-145, has been shown to partially reverse MDSC-mediated immunosuppression. This inhibition enhanced the clinical success of ICIs for head and neck malignancies [75]. However, high doses of IPI-145 can inadvertently inhibit antitumor T cell function, thereby negating the therapeutic benefits of anti-PD-L1 therapy. Moreover, PMN-MDSCs acquire their suppressive function through the upregulation of G-CSF, which is controlled by PI3K activation and regulated by interferon signaling. Interestingly, blocking PI3K or enhancing IFN-I signaling has been demonstrated to lower tumor growth by impairing the immunosuppressive function of MDSCs [34]. One challenge of PI3K inhibition is its potential to inadvertently impair T cell function. This highlights the need for further research into isoform-specific PI3K inhibitors to achieve better clinical outcomes with fewer off-target effects. Interestingly, artemisinin (ART), an antimalarial drug, has been shown to inhibit MDSC recruitment and function in breast cancer models, thereby enhancing the efficacy of anti-PD-L1 therapy both in vitro and in vivo. ART functions by blocking PI3K/AKT/mTOR signaling, which leads to reduced MDSC accumulation and increased T-cell infiltration into tumors [76]. Lastly, it is important to exercise caution when interpreting these findings, as tumors are known to promote MDSC expansion. Therefore, the observed reduction in MDSCs following treatment with a PI3Kδ inhibitor may be an indirect consequence of improved tumor control rather than a direct inhibitory effect on MDSCs. Furthermore, Tregs are also capable of driving MDSC expansion, and a decrease in MDSCs might reflect Treg inhibition rather than a direct action of the PI3Kδ inhibitor on MDSCs.
SIRT 1Silent information regulator 1 (SIRT1) is a histone deacetylase that relies on NAD+ for regulating various biological functions, including cell proliferation, gene regulation, cellular viability, stress resilience, aging, programmed cell death, and metabolic processes [77]. It has been observed to deacetylate p53 and inhibit its transcriptional activity. Therefore, SIRT1 is considered a crucial epigenetic checkpoint in the reprogramming of MDSCs [78]. Numerous studies highlight its importance in controlling MDSC differentiation into the tumor-suppressive M1 subtype, facilitated by glycolytic reprogramming dependent on the mTOR/HIF-1α pathway [77]. Furthermore, SIRT1 plays a role in immunological modulation and metabolic reprogramming by deacetylating transcription factors like HIF1α. Targeting SIRT1 in MDSCs has been shown to regulate the production of TNFα and TGFβ1, aiding in the antigen-specific differentiation of TH1 and iTreg cells [77]. Additionally, SIRT1 acts as a checkpoint in MDSC differentiation into M1 or M2 lineages [79]. Interestingly, SIRT1 deficiency in MDSCs promotes a switch to the M1 lineage, reducing their immunosuppressive capacity and promoting a pro-inflammatory phenotype associated with attacking tumor cells. Furthermore, SIRT1 regulates T-cell-mediated immunity by preventing CD4+ T cells from differentiating into interleukin-9-secreting cells and downregulating transcription factors like NF-κB and AP-1, crucial for regulating T-cell responses [80]. These evidences highlights the critical role of SIRT1 in regulating MDSC activity and maturation, as well as its influence on T-cell immunity. Targeting SIRT1 presents a promising strategy for cancer immunotherapies aimed at overcoming MDSC-mediated immune suppression. Future research should focus on elucidating the precise mechanisms by which SIRT1 modulates MDSC function and identifying synergistic approaches t
Comments (0)