Cell culture
The human pancreatic cancer cell lines PANC-1, MiaPaCa-2, and BxPC-3 were obtained from the American Type Culture Collection, Manassas, VA, USA. These cell lines were cultured under standard conditions at 37°C with 5% CO2 in a humidified incubator. PANC-1 and MiaPaCa-2 cells were maintained in high-glucose DMEM, while BxPC-3 cells were cultured in RPMI 1640 medium. All media were supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, 1% L-glutamine, and 1% nonessential amino acids (Thermo Fisher Scientific). The medium for MiaPaCa-2 was additionally supplemented with 2.5% horse serum. The cells were regularly monitored for mycoplasma infection.
Establishment of RRM1-T734A knock-in of PANC-1 cells
To generate an RRM1-T734A knock-in within PANC-1 cells, we designed a comprehensive CRISPR-Cas9 plasmid. This plasmid, developed by the National RNAi Core Facility in Taipei, Taiwan, contains a lentivirus backbone with sgRNA, Cas9, EGFP, and pPuro. The donor template, an oligonucleotide synthesized by Mission Biotech, has the following sequence:
5′-CTTATTTAGAGTGAACTGGATTGGATTAGCTGCTGGTCTCGTCCTTAAATAATA CATCCCAGCCTTAAGACCCTACAAGGAAAGTAAAGACAATTATTAG-3′.
To introduce the all-in-one plasmid and donor template into PANC-1 parental cells, we employed LT1 reagent for transfection, followed by cell sorting based on GFP expression. For single colony selection, we used 96-well dishes containing GFP-positive cells, followed by puromycin selection and serial dilution in each dish. Each resulting colony was isolated for confirmation of knock-in through PCR and sequencing. The PCR primers used were as follows: Forward primer: 5′-CGGCTGGACAGGAATGTATTT-3′ Reverse primer: 5′-CAGCAAAGCCTTACCACCTC-3′.
Plasmids
The lentiviral shRNA expression vectors pLKO.1-shLacZ, shTK1#1 (TRCN0000010135), shTK1#2 (TRCN0000318728) and shOGT (TRCN0000035064) were obtained from the National RNAi Core Facility (Taipei, Taiwan). Additionally, we purchased the C-GFPSpark-tag Human Thymidine Phosphorylase lentiviral cDNA (I-HG10432-ACGLN) and the lentiviral C-GFPSpark tag vector from Sino Biological.
Establishment of stable cell lines
To establish stable cell lines expressing LacZshRNA, TK1shRNA and OGTshRNA, we infected PANC-1 cells with lentivirus carrying the specified shRNA and then subjected them to selection using 1 μg/mL puromycin. For the creation of a stable cell line expressing the C-GFPSpark-tag vector, or TP, PANC-1 cells were infected and subsequently sorted based on GFP expression using flow cytometry.
Immunoprecipitation of RRM1 and O-GlcNAcylation detection
For RRM1 immunoprecipitation, we lysed cells using IP lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP40, 50 mM NaF, 1 mM Na2VO3, 1 mM DTT, 0.1% SDS, and 2 mM PMSF. We also included phosphatase inhibitors and protease inhibitors. Protein concentrations were determined using the Bradford assay (Bio-rad, Cat# 5000006) and normalized to 1 mg/mL. Subsequently, 500 μg of protein lysate was incubated with either 1 μg of anti-RRM1 antibody (Santa Cruz, Cat# sc-11733) or 1 μg of anti-goat-IgG antibody (Santa Cruz, Cat# sc-2028) at 4 °C overnight. Afterward, we incubated the lysates with protein G beads (Genetex) at 4 °C for 1 h. Following three washes with IP lysis buffer, we eluted the proteins using Laemmli sample buffer for subsequent Western blot analysis. O-GlcNAcylation detection was performed using the O-linked N-Acetylglucosamine (RL2) antibody (Abcam, Cat# ab2739) and the anti-O-GlcNAc (CTD110.6) antibody (Cell Signaling Technology, Cat# 9875).
Immunofluorescence staining
To conduct foci staining of γH2AX, 53BP1, and RPA, we seeded 5000 cells per well in 96-well dishes overnight. The cells were then fixed and permeabilized using 100% methanol for 15 min at −20 °C. After fixation, they underwent three 5 min washes with 0.1% Triton X-100 in TBS buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) on a shaker at room temperature. The cells were blocked with MAXblockTM (Active Motif, Cat# 15252) for 1 h at 37°C and then stained overnight at 4°C with anti-γH2AX (1:1000, Merck Millipore, Cat# 05-636), anti-53BP1 (1:1,000, Merck Millipore Cat# MAB3802), or anti-RPA (1:500, Abcam, Cat# ab109084) antibodies. Following three additional 5 min washes with 0.1% TBST buffer, cells were stained with Alexa Fluor 488 (1:100) or Texas Red (1:200) conjugated secondary antibodies and DAPI (10 mg/ml, 1:1000, Life Technologies) for 1 h at room temperature. Finally, the cells were washed and immersed in PBS for foci analysis using the MD Image Xpress Micro XLS.
Cell proliferation assay
To assess cell proliferation, we seeded 5000 cells per well in a 96-well dish containing 100 μl of growth medium, supplemented with an additional 20 μl of growth medium daily. Cell proliferation was monitored over five consecutive days using the Cell Proliferation Kit II (XTT) (Roche, Cat# 11465015001) following the manufacturer’s guidelines. Results were measured using a Thermo Scientific™ Multiskan™ GO Microplate Spectrophotometer, recording absorbance at OD492 and OD690 nm.
Cell death analysis
The viability of PANC-1 cells was assessed using the LIVE/DEAD™ Cell Imaging Kit (488/570) (Invitrogen, Cat# R37601). A 2X stock solution containing live green and dead red reagents was prepared and mixed with PBS. This mixture was incubated with the cells at room temperature for 15 min, along with Hoechst 33342 to stain the cell nuclei. Fluorescence imaging was conducted to classify cells as viable (green fluorescence) or non-viable (red fluorescence).
Cell cycle synchronization and analysis
To synchronize the cell cycle, cells were exposed to 100 ng/mL of nocodazole (Sigma-Aldrich) for 16 h, followed by two PBS washes. Cells were then plated in culture media for 6, 12, 18, and 24 h. Approximately 1 million cells were fixed with 70% cold ethanol and stored at −20 °C overnight. The fixed cells were centrifuged at 2000 rpm for 5 min, and the supernatant was removed. After washing with cold PBS, a PI staining buffer (0.04 mg/mL Propidium Iodide, 2 mg RNase A, and 0.1% Triton X-100) was applied and incubated at room temperature for 30 min. Stained cells were analyzed using a flow cytometer.
Soft agar colony formation assay
For the soft agar colony formation assay, 500 cells were seeded in a 0.35% agar layer over a 0.5% agar layer in 12-well dishes. The cells were cultured in a humidified 37 °C incubator for 9 days. Colony-forming efficiency was assessed using light microscopy, counting colonies with diameters exceeding 50 μm.
Sphere formation assay
The sphere formation assay involved seeding 5000 cells per well in a low-attachment 6-well dish, incubating for 6 days. Spheroids larger than 100 μm in diameter were counted and analyzed using light microscopy.
Western blot analysis
Cells were lysed using RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 50 mM NaF, 1% Nonidet P-40) supplemented with a protease inhibitor cocktail and a phosphatase inhibitor cocktail. Equal amounts of protein lysates were separated via SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% milk in TBST (0.1% Triton X-100) at room temperature for 1 h, then incubated with primary antibodies overnight at 4 °C. After incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h, signals were visualized using Immobilon Forte Western HRP Substrate (Merck Millipore, Cat# WBLUF0500) and captured using the UVP BioSpectrum 500 Imaging System. Gel-Pro software was used to quantify the intensity of each band in the immunoblotting.
Animal protocol
All animal experiments were approved by the Institutional Animal Care and Utilization Committee of Academia Sinica, Taipei, Taiwan (IACUC#20-03-1451 and IACUC#23-11-2084). Mice were housed in a specific pathogen-free facility at 20 °C ± 2 °C with a 12/12 h light/dark cycle and were given unrestricted access to water and standard laboratory chow.
Tumor growth in mice
To create GFP-Luc PANC-1 cells, PANC-1 cells were infected with a GFP-Luciferase lentivirus and subsequently sorted for GFP expression via flow cytometry. NOD/SCID/IL2Rγnull (NSG) mice, obtained from Jackson Laboratory, were bred in-house at the Academia Sinica animal core facility. Eight-week-old male NSG mice of similar body weights were selected for the experiments. In the subcutaneous tumorigenicity assay, 10 million PANC-1 cells were mixed with 50 μl of Matrigel and injected subcutaneously into the right flanks of NSG mice. Tumor size and body weight were monitored weekly. Seven weeks post-implantation, mice were euthanized, and their tissues were collected. Tumor volume was calculated using the formula: tumor volume = 1/2 × width2 × length. For the orthotopic tumorigenicity assay, 5 × 106 cells in 30 μl of Matrigel were injected into the pancreas of NSG mice. Two months after orthotopic implantation, mice were euthanized, and tumor size and weight were measured. The metastases of PANC-1 cells to liver and lung metastases was assessed using the IVIS kinetics imaging system (Caliper LifeSciences).
NTP and dNTP quantification
Total 1 × 107 cells were suspended in 1 ml of 80% cold methanol ( − 20 °C), then sonicated and centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant was transferred to a new tube and dried using a SpeedVac (LaboGene). The pellets were reconstituted in 1 ml of ddH2O, mixed with 3.75 ml of cold chloroform/methanol (1:1), and agitated using an ELMI Intelli-mixer ERM-2 for 10 to 15 min at 4 °C. Next, 1.25 ml of cold chloroform (4 °C) was added, vortexed for 1 min, followed by 1.25 ml of ice-cold ddH2O, and further vortexedfor 1 min. The sample was stored at −80 °C for 25 min, then centrifuged at 13,000 rpm for 10 min at 4 °C. The upper hydrophilic layer was collected into a new tube and dried using a SpeedVac for LC-ESI-MS detection.
Nucleoside metabolite analysis
Total 5 × 106 cells were harvested and washed with chilled PBS. The pellets were reconstituted in a solution of methanol: acetonitrile: ddH2O (2:2:1) and sonicated three times for 30 s each. After centrifugation at 12,000 rpm for 10 min at 4 °C, the supernatant was collected and dried using a SpeedVec (LaboGene). The dried samples were reconstituted in 50% methanol ( − 20 °C) for LC-ESI-MS detection.
LC-ESI-MS for metabolomics detection
To amplify signals of carboxylic acid and organic phosphate, 5 µl of aniline/HCl reaction buffer (0.3 M aniline [Sigma-Aldrich, USA] in 60 mM HCl) and 5 µl of 20 mg/ml N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC; Sigma-Aldrich, USA) were added to each hemocyte residue sample. After vortexing, the mixtures were incubated at 25 °C for 2 h. The reaction was halted by adding 5 µl of 10% ammonium hydroxide. The aniline-derivatized samples were analyzed using an LC-ESI-MS system consisting of an ultra-performance liquid chromatography system (Ultimate3000 RSLC, Dionex) and a quadrupole time-of-flight mass spectrometer with an electrospray ionization (ESI) source (maXis UHR-QToF system, Bruker Daltonics). Metabolites were separated via reversed-phase liquid chromatography on a BEH C18 column (2.1 × 100 mm, Waters). The LC settings were autosampler temperature at 4 °C, injection volume of 10 µl, and flow rate at 0.4 mL/min. The elution began with 1% mobile phase B (0.1% formic acid in ACN) for 4 min, followed by a gradient from 99% mobile phase A (0.1% formic acid in ddH2O) and 1% mobile phase B. After being held at 1% for 0.5 min and increased to 60% over 5 min, mobile phase B was further elevated to 90% in an additional 0.5 min, maintained at 90% for 1.5 min, and then reduced back to 1% in 0.5 min. Subsequently, the column was re-equilibrated by pumping 99% B for 4 min. The LC-ESI-MS chromatograms were acquired under these conditions: dry gas temperature at 190 °C, dry gas flow rate at 8 L/min, nebulizer gas at 1.4 bar, and capillary voltage at 3500 V. Mass spectra in the negative ion mode were recorded across the range of m/z 100–1000. Data acquisition was done using HyStar and microTOF control software (Bruker Daltonics), and analyzed with DataAnalysis and TargetAnalysis software (Bruker Daltonics). Each metabolite was identified by matching its theoretical m/z value and isotope pattern derived from its chemical formula. Quantification was performed by summing the corresponding area of the extracted ion chromatogram.
RNA extraction and qPCR analysis
Total RNA was extracted from the cells using the TRIzol method (Life Technologies, 15596018) according to the manufacturer’s instructions. The isolated mRNA was reverse-transcribed into cDNA using the Maxima First Strand cDNA Synthesis Kit from Thermo Fisher Scientific Inc. Gene expression levels were quantified by real-time PCR with the SYBR FAST qPCR Kit (KAPA Biosystems, Wilmington, MA, USA) on a StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). GAPDH was used as the internal control for normalizing mRNA expression. Fold changes were calculated using the 2-ΔΔCt method after normalization with the internal control. The sequences of the QPCR primers for specific genes were as follows:
TK1 forward primer: 5’-CCTTCCTGCCACTGCCGCCTACTG-3’, TK1 reverse primer: 5’-TCCACGCCTCCCGACTTCCTCCTG-3’; TP forward primer: 5’-CTGGAGTTATTCCTGGATTCA-3’; TP reverse primer: 5’-TCTGCTCTGGGCTCTGGATGA-3’; GAPDH forward primer: 5’-GCTGCAGGGCCTCACTCCTTTT-3’; GAPDH reverse primer: 5’-AGAGCCAGTCTCTGGCCCC-3’.
TK1 stabilization analysis
PANC-1 cells were seeded in a 6-well dish and allowed to adhere overnight. These cells were treated with 50 μg/ml cycloheximide (CHX, Sigma, Cat# C1988) for 0, 2, 4, and 8 h, then collected using RIPA buffer for Western blot analysis.
Cell cytotoxicity assay
A total 5 × 103 cells were plated per well in 96-well dishes and exposed to varying concentrations of gemcitabine (0–100 μM) for 3 days. Cell viability was assessed using the Cell Proliferation Kit II (XTT) (Product No. 11465015001) according to the manufacturer’s protocol. The IC50 value was calculated using GraphPad Prism 9 software.
Statistical analysis
The data were presented as means ± SD, as detailed in the figure legends. A two-tailed Student’s t-test was used to compare differences between the control and experimental groups. * denotes statistical significance with p < 0.05, ** indicates p < 0.01, *** represents p < 0.001 and **** represents p < 0.0001. The specific n numbers used in the respective sections were described in the figure legends.

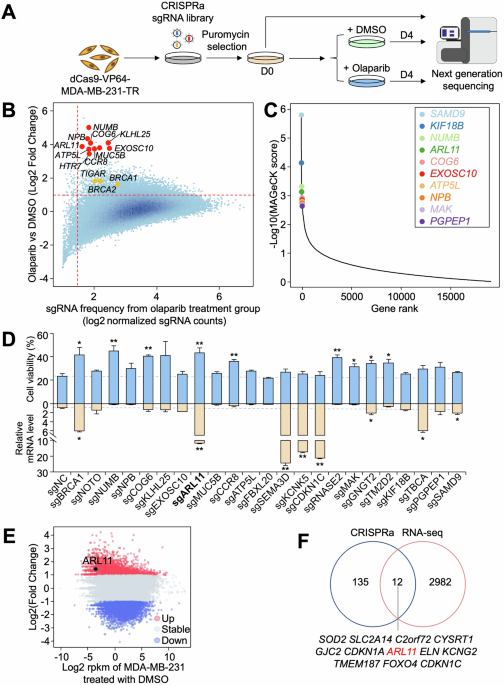
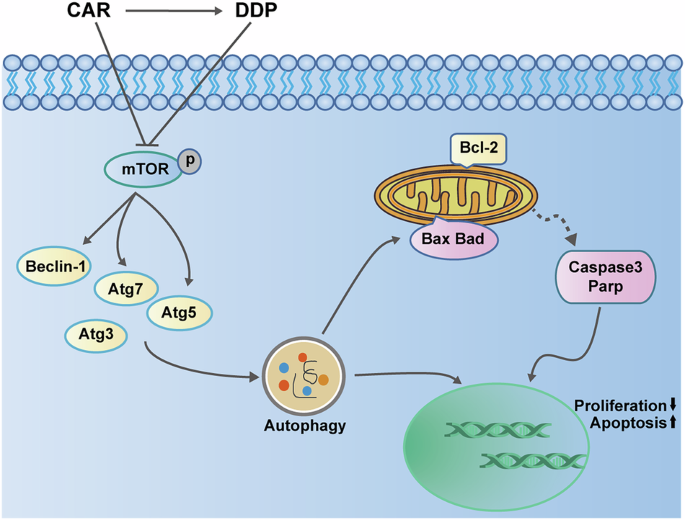
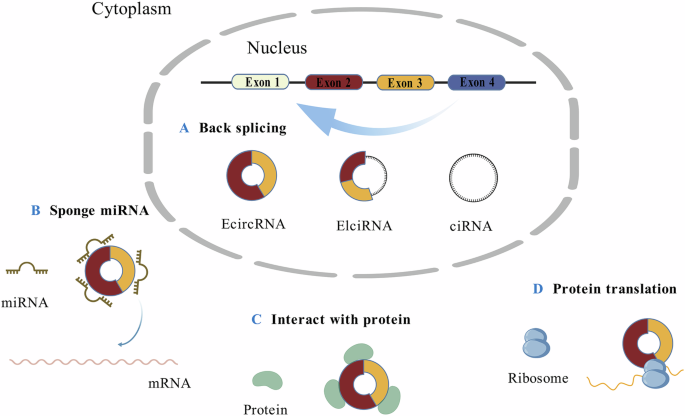




Comments (0)