
Remember me

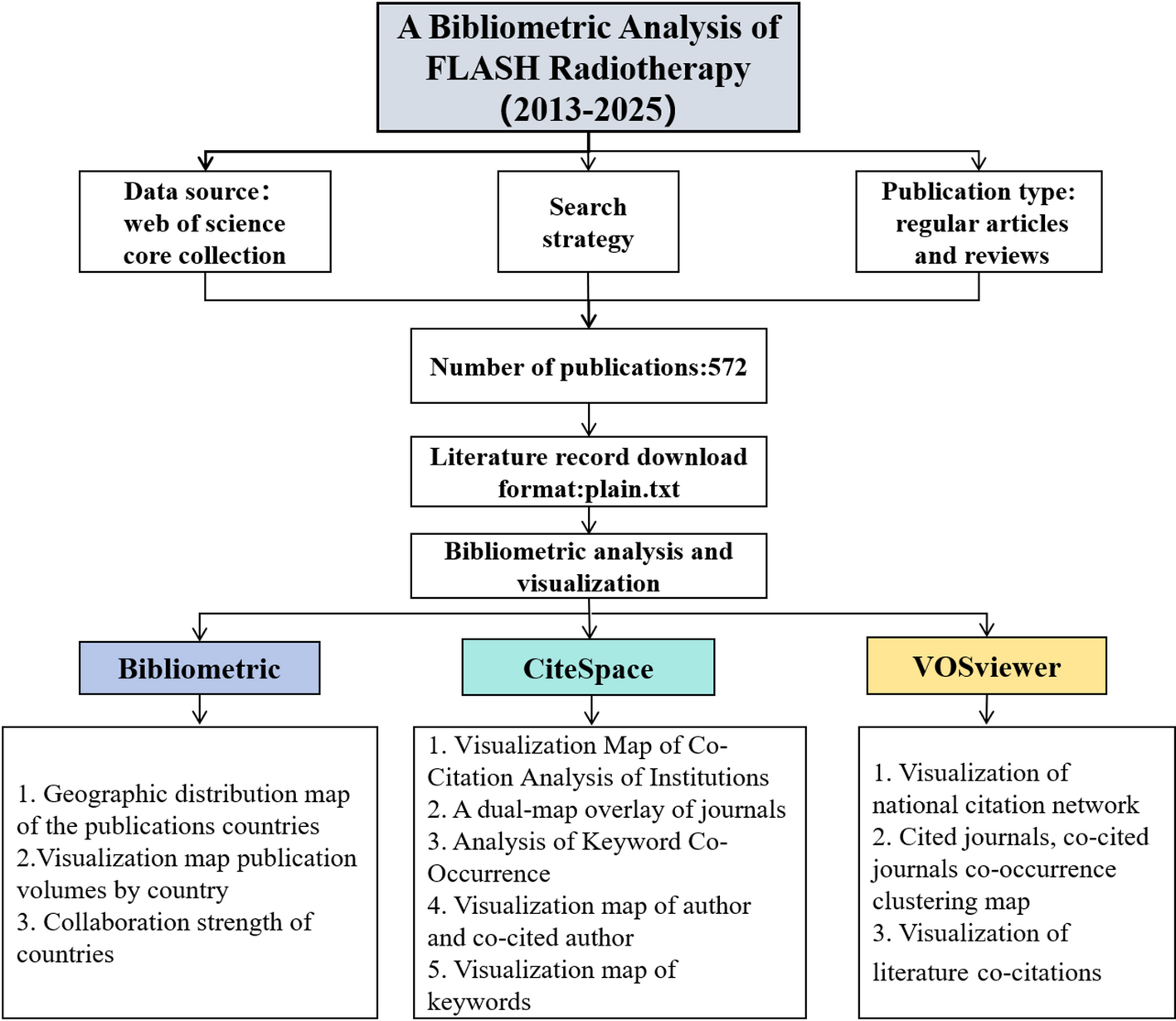


The processing flow of this study is shown in Fig. 1.
Fig. 1
Schematic workflow illustrating the data analysis process
All methods were carried out in accordance with relevant guidelines and regulations.
2.1 Data acquisition and standardizationClinical information and RNA sequencing data were obtained from the publicly accessible FU-iCCA cohort database [13]. The criteria for inclusion were as follows: (a) samples confirmed as iCCA by surgical pathology; (b) samples accompanied by relevant clinical information and a gene expression matrix; and (c) samples with complete follow-up data, including survival duration, survival status. The exclusion criterion was: (a) samples that did not have comprehensive clinical information. Additionally, an external validation cohort was formed by combining 30 iCCA cases with RNA-seq and follow-up data from the TCGA-CHOL dataset and 30 iCCA cases with RNA-seq and follow-up data from GSE107943 [27].
In total, 244 samples were included in the analysis. The cohort comprised 106 (43.44%) female and 138 (56.56%) male patients, with a median age of 60.58 years (range: 27–86). Among them, 15 (6.15%) patients had a history of liver fluke infection, while 10 (4.10%) had biliary tract stone disease. Additionally, 50 (20.49%) patients exhibited lymph node metastasis (LNM), and 49 (20.08%) had perineural invasion. The presence of vascular invasion was observed in 103 (42.21%) cases, whereas liver cirrhosis was diagnosed in 22 (9.02%) patients.
Furthermore, the cohort was randomly divided into a training cohort and a testing cohort in a 7:3 ratio, utilizing R version 4.4.0 and Zstats 1.0 (www.zstats.net). The baseline clinical characteristics of both the training and testing cohorts are summarized in Table 3.
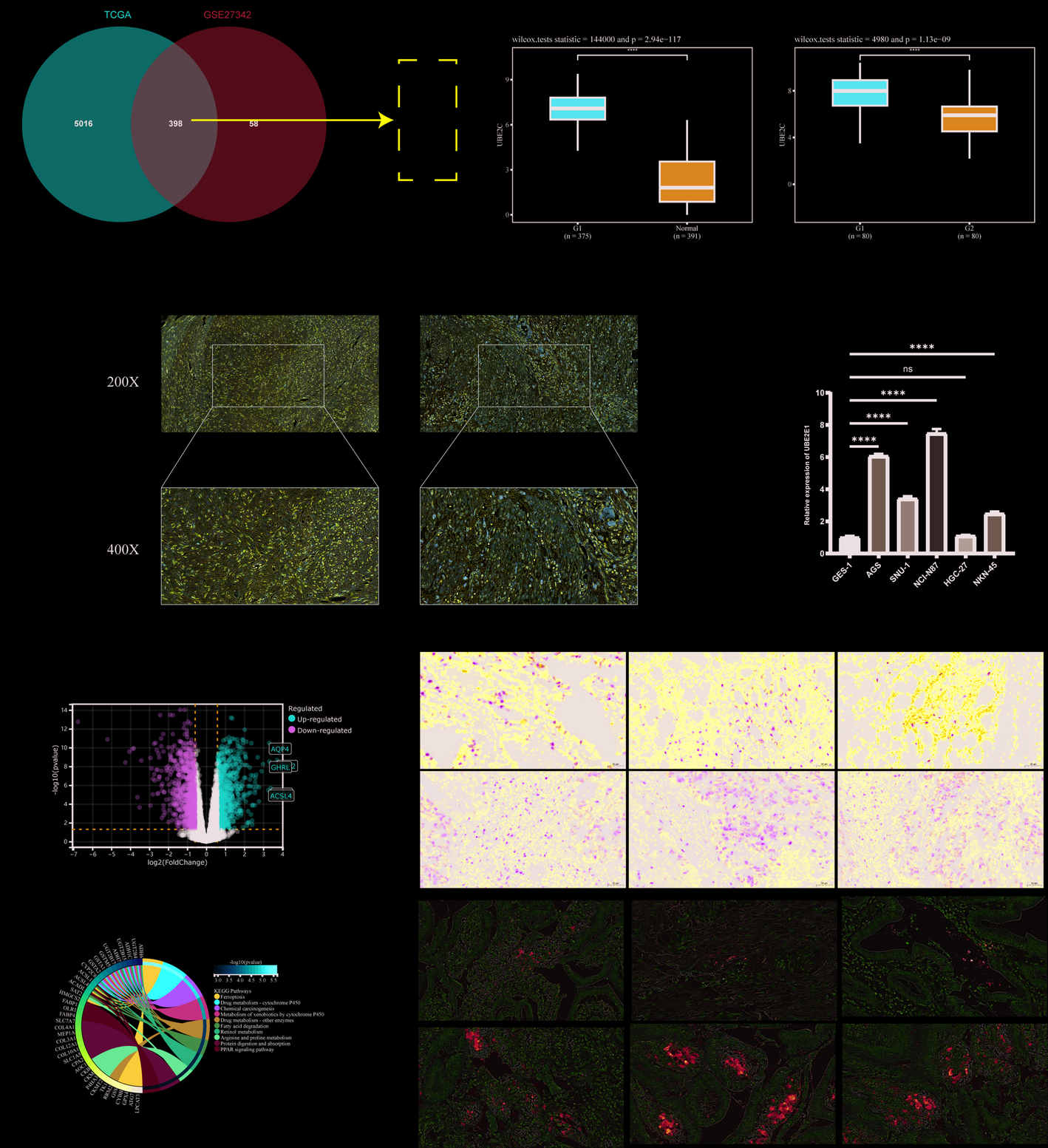
2.2 Screening of overall survival (OS) prognostic genesWe obtained 339 human glycolysis-related genes from the Molecular Signatures Database (MSigDB) database, including the BIOCARTA Glycolysis Pathway, GO Glycolytic Process, Hallmark Glycolysis, KEGG Glycolysis and Gluconeogenesis, and Reactome Glycolysis and extracted glycolysis-related genes from the iCCA dataset within the FU-iCCA cohort. Subsequently, we utilized the survival package in R to conduct a univariate Cox regression analysis, identifying 121 glycolysis-related genes that were significantly associated with OS in iCCA patients (p < 0.05).
2.3 Identification of molecular subgroupsCluster analysis was performed using ConsensusClusterPlus [28]. Agglomerative partitioning around medoids (PAM) clustering with 1-Pearson correlation distances was employed. Resampling was performed on 80% of the samples for 10 repetitions. The optimal number of clusters was determined by evaluating the cumulative distribution function (CDF) plot. Specifically, the number of clusters (K) was selected by examining the area under the CDF curve and the Delta CDF, which measures the relative change in the CDF as K increases. To ensure robust clustering results, the optimal K value was chosen based on a balance between a large area under the CDF curve and the slowest rate of decline in the Delta CDF. Additionally, internal consistency was considered, with the highest cluster consistency observed at K = 2 and the second-highest at K = 7.
2.4 Functional analysesDifferentially expressed genes (DEGs) between the two clusters were identified using the R package limma (version 3.40.6) [29], with|log2FC| ≥ 1 and FDR < 0.01 as the selection criteria. For Gene Ontology (GO) enrichment analysis, three subcategories were analyzed: Biological Process (GOBP), Cellular Component (GOCC), and Molecular Function (GOMF). The corresponding gene sets (c5.go.bp.v7.4.symbols.gmt, c5.go.cc.v7.4.symbols.gmt, c5.go.mf.v7.4.symbols.gmt) were downloaded from the Molecular Signatures Database (MSigDB) [30] and analyzed using clusterProfiler (version 3.14.3) [28]. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was conducted using the latest KEGG pathway gene annotations retrieved via the KEGG REST API, followed by enrichment analysis with clusterProfiler. For Gene Set Variation Analysis (GSVA), the R package GSVA (DOI:https://doi.org/10.18129/B9.bioc.GSVA, version 1.40.1) was used to calculate enrichment scores for each sample. Gene ranking was based on expression profiles using the method by Hänzelmann et al. [31]. The c5.go.bp.v7.4.symbols.gmt gene set was obtained from the MSigDB and was used to assess pathways and molecular mechanisms. Gene sets containing 5 to 5000 genes were considered. Differential analysis of enrichment scores was conducted using the R package limma (version 3.40.6) [29]. The lmFit function performed multiple linear regression, followed by eBayes to compute moderated statistics and identify significantly enriched pathways between groups. For Gene Set Enrichment Analysis (GSEA), the GSEA software (version 3.0) from the Broad Institute [32] was used to group samples based on molecular subgroups. The h.all.v7.4.symbols.gmt gene set was downloaded from the MSigDB to assess relevant pathways and molecular mechanisms. Using gene expression profiles and phenotype grouping, the minimum gene set size was set to 5 and the maximum to 5000, with 1000 resampling iterations. Pathways with p < 0.05 and FDR < 0.25 were considered statistically significant.
2.5 Establishment and validation of risk modelIn this section, we utilized the R package glmnet to integrate survival time, survival status, and gene expression data, applying the Lasso-Cox method for regression analysis. Additionally, we implemented 10-fold cross-validation to optimize the model. We set the lambda value to 0.108289354101911, ultimately identifying nine genes. The risk score for each patient in the training, testing and validation cohorts was calculated using the following formula: Risk Score = − 0.00430195586928747×ALDH1B1 + 0.000608847034696516×DDIT4 + 0.00430380061449737×GALE + 0.00151186854634352×HK1 + 0.0103354979163438×HMMR + 0.00132273347738233×PGAM1 + 0.00252631748191928×PGK1 + 0.00306647381422908×PLOD1 + 0.0101780840135645×SAP30. Patients were subsequently categorized into high-risk and low-risk groups. Receiver Operating Characteristic (ROC) analysis was conducted to evaluate the predictive efficiency of the model.
2.6 Nomogram model constructionInitially, single- and multivariate Cox modeling analyses were performed using R version 4.4.0 (2024-04-24) and Zstats 1.0 (www.zstats.net). Based on the results of the multivariate Cox proportional hazards analysis, we utilized the R package rms to integrate survival time, survival status, and four features. A nomogram was constructed using the Cox method to assess the prognostic significance of these features in a cohort of 170 samples. The overall C-index of the model was 0.793, with a 95% CI of 0.744–0.842, and a p-value of 1.96e-31.
2.7 Specimens and cell cultureAll human tissue samples were obtained from Zhongshan Hospital, Fudan University, following informed consent and ethical approval. A total of 80 iCCA tumor tissue specimens were collected for the construction of a tissue microarray (TMA), among which paired tumor and adjacent normal liver tissues were available from four patients for RNA extraction and subsequent qRT-PCR analysis. The inclusion criteria were as follows: (1) Histopathologically confirmed primary iCCA cases that underwent curative-intent surgical resection at Zhongshan Hospital, Fudan University, between June 2018 and December 2023. (2) No prior history of neoadjuvant or other anti-tumor therapies before surgery. The exclusion criteria included: (1) Incomplete clinicopathological data or follow-up information. (2) Concurrent diagnosis of other malignancies. All pathological diagnoses were independently reviewed and confirmed by at least two senior pathologists from Zhongshan Hospital, Fudan University.
The RBE and HuCC-T1 cell lines, both of which are human-derived iCCA cell lines, were obtained from the Liver Cancer Institute, Zhongshan Hospital. All cells were cultured in RPMI-1640 medium (Sigma-Aldrich R8758), supplemented with 10% fetal bovine serum (Hyclone SH30406.05) and 1% penicillin-streptomycin (NCM Biotech C125C5). The cells were maintained in a 37 °C incubator with 5% CO₂. The Research Ethics Committee of Zhongshan Hospital approved the study, and written informed consent was obtained from all patients.
2.8 RNA extraction and qRT-PCRTotal RNA was isolated from the tissues and cells using TRIzol reagent (Invitrogen 15596018CN) following the manufacturer’s guidelines. The isolated RNA was then reverse transcribed using the HiScript III RT SuperMix for qPCR (+ gDNA wiper) kit (Vazyme R323). For quantitative PCR analysis, ChamQ Universal SYBR qPCR Master Mix (Vazyme Q711) was utilized. β-actin acted as the internal control for normalization, and relative expression levels were assessed using the 2−ΔΔCt method.
2.9 Immunohistochemistry (IHC) stainingThe iCCA tissue microarray chips constructed by our research group were stained with anti-HMMR antibody (1:100, Proteintech 15820-1-AP) to assess the expression levels of HMMR. The Immunohistochemical (IHC) score was calculated using the Immunoreactive Score (IRS) method, which involves multiplying the staining intensity score (negative = 0, weak = 1, moderate = 2, strong = 3) by the positive rate score (negative = 0, 1–25% = 1, 26–50% = 2, 51–75% = 3, 76–100% = 4). Tumor sections with an IHC score exceeding 8 points were classified as the “high expression” group.
2.10 RNA interference and cell transfectionThe siRNA targeting HMMR and a control siRNA were designed and synthesized by Shanghai Xinzhuo Biotechnology Co., Ltd. The sequences are as follows: 5’-GGACCAGUAUCCUUUCAGATT-3’ (siHMMR_1), 5’-GACCAGGACUAAUGAACUATT-3’ (siHMMR_2), and 5’-UUCUCCGAACGUGUCACGUTT-3’ (siCtrl). Following the manufacturer’s instructions, RBE and HuCC-T1 cells were transfected using Lipofectamine 3000 reagent (Invitrogen, L3000015), with siCtrl transfection used as a control. After 24 h, the expression levels of HMMR in RBE and HuCC-T1 cells were assessed using qRT-PCR.
2.11 Cell migration and invasion assay1–5 × 104 overnight-starved cells were seeded into the upper chamber (Corning 3422) for the invasion assay (with Basement Membrane Matrigel, Corning, 356230) or the migration assay (without Matrigel). RPMI-1640 supplemented with or without 10% FBS was added to the lower and upper chambers, respectively. After 36 to 48 h of incubation, cells were fixed with 4% paraformaldehyde. Subsequently, the cells were stained with 0.1% crystal violet solution and counted manually.
2.12 Cell proliferation assayA total of 3 × 103 cells per well were seeded into a 96-well plate and cultured in RPMI-1640 supplemented with 10% FBS. Absorbance at 450 nm was measured at 6, 36, 60, 84 and 110 h. One hour before each measurement, the culture medium was replaced with fresh RPMI-1640 and 10% CCK-8 to ensure accurate readings.
2.13 Statistical analysesStatistical analyses and visualizations were conducted using R version 4.4.0, Sangerbox 3.0 (http://www.sangerbox.com/tool), and Zstats v1.0 (www.zstats.net). Survival analysis was conducted using the Kaplan-Meier method, as well as univariate and multivariate Cox regression models. The predictive performance of the risk model was evaluated through ROC analysis using the R package pROC (version 1.17.0.1), calculating the area under the curve (AUC). Subgroup analysis was performed by stratifying patients based on clinical information. Categorical data are presented as counts and percentages, whereas continuous data are expressed as means ± standard deviation (SD). The Student’s t-test was used to compare two groups, and one-way ANOVA was employed for comparisons involving three or more groups. A P value < 0.05 was considered statistically significant.
Comments (0)