
Remember me



This study developed a CCA prognostic risk model based on bioinformatics analysis, utilizing data from the TCGA, GEO, and EMBL-EBI databases. Data from the TCGA and GEO databases were used as the training set, while data from the EMBL-EBI database served as the validation set. The model was established thr ough the screening of DEGs, univariate Cox regression, and LASSO regression analysis. The model was subsequently validated using the validation set. Further gene enrichment analysis and immune infiltration analysis were conducted to enhance the evaluation of CCA patient prognosis and to aid in the formulation of targeted clinical treatment strategies. The overall analytical workflow is presented in Fig. 1.
Fig. 1
Overall Analysis Workflow for Constructing a CCA Prognostic Risk Model Based on TCGA and GEO Data Screening of OSRGs
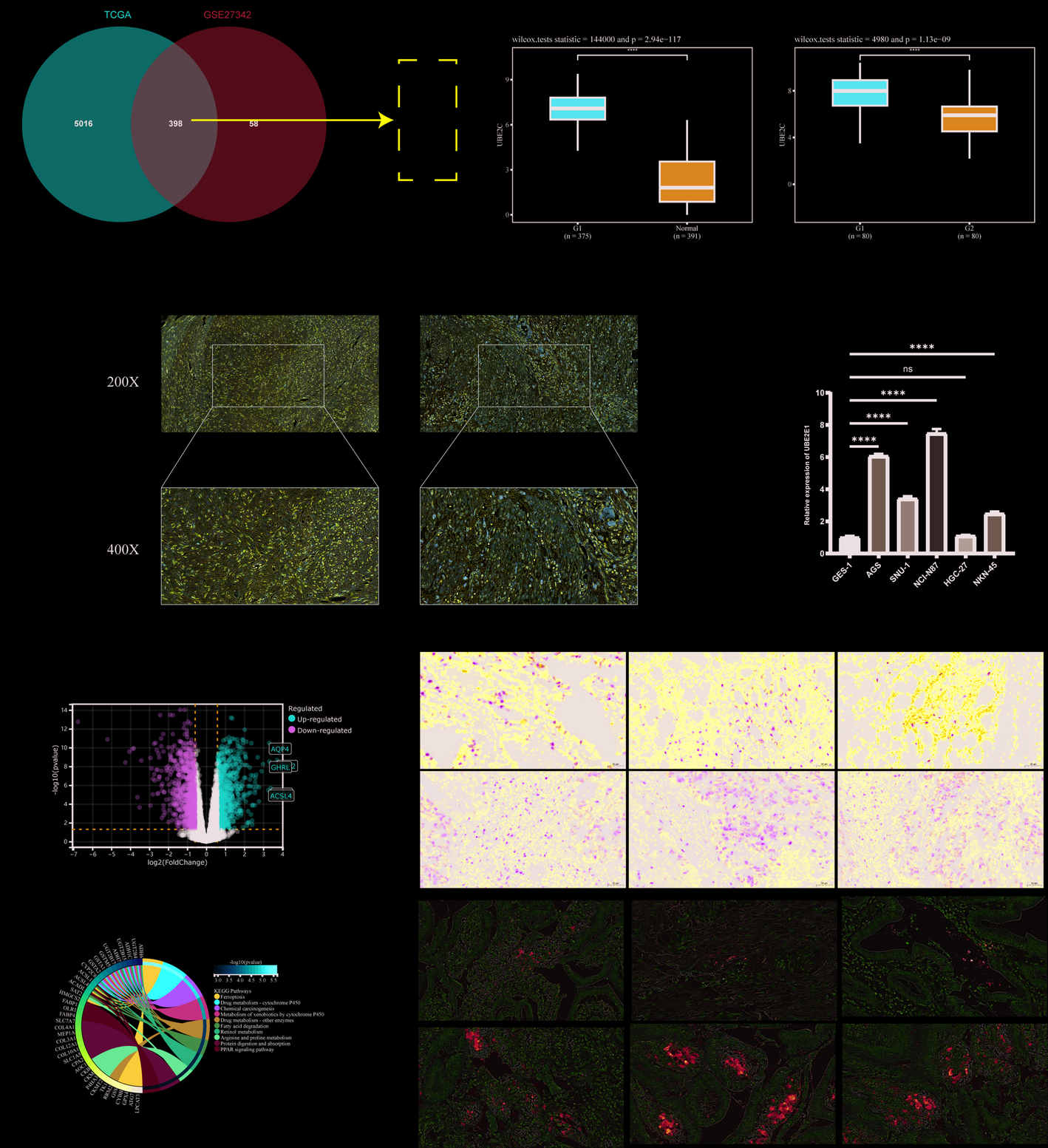
3.2 Screening of DEGs in TCGA and GEO datasetsA total of 66 tumor samples and 36 control samples from CCA patients were extracted from the TCGA and GEO databases. Batch effects were corrected using the ComBat function in the “sva” R package (Fig. 2A-B). Using the “limma” package, we performed a comparative analysis on the batch effect-corrected CCA and control samples, identifying 3,632 DEGs, with 1,991 genes downregulated and 1,641 genes upregulated (Fig. 2C-D). We then intersected these 3,632 DEGs with 576 OSRGs retrieved from the GeneCards database, resulting in 122 differentially expressed OSRGs, as illustrated in Fig. 2E (Venn diagram). Further analysis of these 122 genes revealed their differential expression between CCA and normal samples, as well as their expression across various clinicopathological features such as age, sex, and stage (Fig. 2F-G).
Fig. 2
Screening of DEGs in TCGA and GEO Datasets. Note: (A) PCA of the combined TCGA and GEO datasets. (B) PCA of the combined dataset after correction. (C) Volcano plot of DEGs in the dataset. (D) Heatmap of DEGs in the dataset. (E) Venn diagram of the intersection between DEGs and OSRGs. (F-G) Heatmaps of differentially expressed OSRGs
3.3 Selection of prognostic genes and survival analysis in CCAUsing univariate Cox regression analysis, 19 prognostically significant candidate genes (P < 0.05) were identified after multiple testing corrections with the Benjamini-Hochberg method, as shown in Fig. 3A. These include SLC7A11, NAT2, GRIN2A, CP, and NOX4. Among them, the hazard ratios (HR) for RORA and GRIN2A were less than 1, suggesting they might be protective factors for CCA patients’ prognosis. In contrast, the remaining 17 genes, such as CP, CDK1, CCNB1, and PLAU, had HRs greater than 1, indicating they may be risk factors affecting CCA prognosis. These 19 candidate genes were then subjected to LASSO regression analysis (Fig. 3B-C) for further dimension reduction and prognostic gene selection. The optimal Lambda value (-2.810403) was selected using cross-validation to achieve the best model. A total of 10 prognostic genes were identified as potential biomarkers for CCA patients. These genes include CP, RORA, NOX4, BIRC5, MT2A, LCN2, XDH, MBL2, CDK1, and GRIN2A (Fig. 3D-E). Subsequently, survival correlation analysis of these 10 prognostic genes was performed, and K-M survival curves were generated. As shown in Fig. 3F, RORA and GRIN2A are associated with improved survival, while the other eight genes correlate with poorer outcomes, suggesting their roles as protective and risk factors in CCA prognosis.
Fig. 3
Screening and Survival Analysis of CCA Prognosis-Related Genes. Note: (A) Forest plot of univariate Cox regression analysis showing genes related to CCA prognosis. (B) Path diagram of regression coefficients using the LASSO regression algorithm. (C) Verification curve of regression coefficients using the LASSO regression algorithm. (D) Correlation circle plot of candidate CCA prognosis-related genes. (E) Interaction network of candidate CCA prognosis-related genes. (F) KM survival curves of CCA prognosis-related genes
3.4 Construction and validation of the CCA prognostic risk modelIn both the training and validation cohorts, we obtained risk scores for all samples. The heatmap illustrated the expression of the 10 prognostic genes, while the scatter plot revealed the survival times of patients in different risk groups. The distribution of risk scores for each patient was shown in the risk score distribution plot. As depicted in Fig. 4A-B, an increase in risk scores was significantly associated with decreased patient survival rates. Based on clinical information such as age, sex, and tumor stage, Fig. 4C-E displays the distribution of these clinical characteristics (such as stage, age, and gender) within the different risk groups.
Fig. 4
Construction of the CCA Prognostic Risk Model. Note: (A) Triple plot of risk scores in the TCGA and GEO training sets. (B) Triple plot of risk scores in the EMBL-EBI validation set. (C) Tumor staging in high- and low-risk groups. (D) Age distribution in high- and low-risk groups. (E) Gender distribution in high- and low-risk groups
To evaluate the predictive ability of the risk model, we analyzed the risk scores from both the training and validation cohorts. The training cohort was divided into high- and low-risk groups based on the median risk score, and KM curves and ROC curves were generated (Fig. 5A-B). The KM curves revealed differences in OS between the high- and low-risk groups, while the ROC curves predicted the AUC values for 1-, 3-, and 5-year OS. The results indicated a significantly higher mortality rate in the high-risk group, with the ROC curves demonstrating good predictive performance. Additionally, PCA and tSNE were used to assess the ability of the risk model to distinguish patients. The two groups exhibited a clustering trend in the reduced-dimensional space, indicating that gene expression patterns correlate with patient risk stratification (Fig. 5C-D).
Fig. 5
Validation of the CCA Prognostic Risk Model. Note: (A) KM survival curve for OS of CCA patients in the TCGA dataset; (B) ROC curve for CCA patients in the TCGA dataset; (C) PCA analysis for CCA patients in the TCGA dataset; (D) t-SNE analysis for CCA patients in the TCGA dataset
3.5 Construction of theprognostic nomogramTo further evaluate the predictive value of the established 10-gene Prognostic Risk Model, we incorporated risk scores and clinicopathological features from the training set into a multivariate Cox regression analysis. Additionally, we constructed a nomogram to illustrate the potential impact of these clinicopathological characteristics and risk scores on the prognosis of CCA patients. The results from the multivariate Cox regression analysis in the TCGA and GEO training set indicated that the risk score was an independent prognostic factor for CCA (HR = 3.97, 95% CI: 0.83–1.93, P < 0.01). Additionally, we generated calibration curves for 1–3 years to validate the model’s predictive accuracy. For the first CCA patient sample, the total score was 191 points. The prediction indicated a very low probability of death within 1–3 years, closely aligning with the actual observed outcomes (Fig. 6A-B).
Fig. 6
Construction of the CCA Prognostic Risk Model Nomogram. Note: (A) Bar plot of risk scores and clinical characteristics of patients based on the dataset. (B) Calibration curves predicting 1-, 2-, and 3-year survival rates for CCA patients
3.6 Enrichment analysis of DEGs in high- and low-risk groupsFunctional and pathway analyses of DEGs were performed to explore the prognostic differences between risk groups. A total of 140 significantly different genes were identified between the two groups, with 29 genes downregulated and 121 genes upregulated, as shown in Fig. 7A-B. These DEGs were subjected to GSEA and GSVA enrichment analyses. The GSEA enrichment analysis revealed the activation of pathways such as coagulation, E2F targets, G2M checkpoint, inflammatory response, mTORC1 signaling, and xenobiotic metabolism in the high-risk group, as depicted in Fig. 7C. The GSVA enrichment results indicated the activation of galactose metabolism, P53 signaling pathway, tyrosine metabolism, PPAR signaling pathway, complement and coagulation cascades, folate metabolism, and folate biosynthesis (Fig. 7D). The GSVA bar chart (Fig. 7E) quantitatively illustrates the significance of these pathways in the high-risk group, with yellow bars representing significantly activated pathways and blue bars representing significantly suppressed pathways.
Fig. 7
Enrichment Analysis of DEGs Between High and Low-Risk Groups. Note: (A) Volcano plot of differentially expressed prognostic genes between high and low-risk groups. (B) Heatmap of differentially expressed prognostic genes between high and low-risk groups. (C) GSEA enrichment analysis of DEGs between high and low-risk groups. (D-E) GSVA enrichment analysis of DEGs between high and low-risk groups
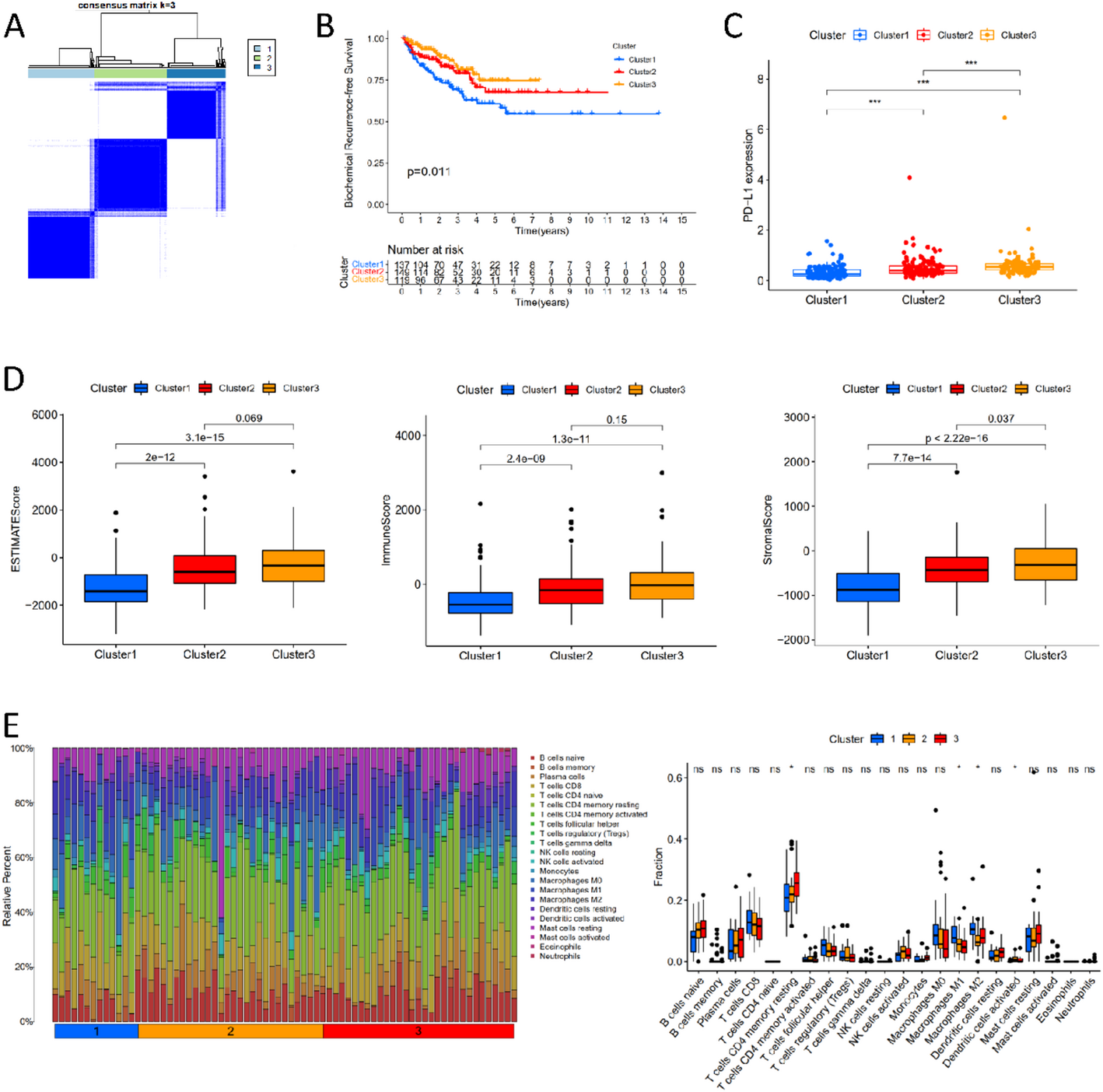
3.7 TME and immune cell infiltration analysisPrevious studies have shown a strong correlation between tumor progression and the TME. An immunosuppressive TME facilitates immune evasion, promoting tumor progression [30]. Additionally, tumor development is closely linked to chronic inflammation, with oxidative stress, ROS, and RNS influencing the TME and driving tumor progression, including CCA [31, 32]. TME analysis compared stromal and immune components between high- and low-risk groups. Using TIMER, stromal and immune scores were calculated for each sample. The high-risk group’s immune and ESTIMATE scores were significantly higher (p < 0.05, Fig. 8A).
Fig. 8
TME and Immune Cell Infiltration Analysis. Note: (A) Stromal scores, immune scores, and ESTIMATE scores of high and low-risk CCA patients. (B) Bar chart of differences in immune cell infiltration between high and low-risk groups. (C) Box plot of differences in immune cell infiltration between high and low-risk groups. (D) Heatmap of the correlation between prognostic genes and immune cell infiltration in CCA. (E) Bar chart of the correlation between risk scores and immune cell infiltration in CCA patients
CIBERSORT analysis revealed significant differences in immune cell infiltration between the two groups (Fig. 8B-C). The high-risk group had higher levels of central memory CD8 T cells, activated CD4 T cells, γδ T cells, type 1 and type 2 helper T cells, regulatory T cells, myeloid-derived suppressor cells, activated dendritic cells, and neutrophils, while eosinophils were more prevalent in the low-risk group.
Correlation analysis between prognostic genes and immune cells showed that BIRC5 and CDK1 were positively correlated with type 2 helper T cells, activated CD4 T cells, and γδ T cells, while GRIN2A was negatively correlated with type 2 helper T cells, γδ T cells, and neutrophils. MT2A was positively correlated with myeloid-derived suppressor cells but negatively correlated with eosinophils (Fig. 8D).
Further analysis of risk scores and immune cells indicated that eosinophils were negatively correlated with risk scores, whereas most other immune cells showed a positive correlation (Fig. 8E).
3.8 Role of risk scores in predicting immunotherapy and other treatmentsTo validate the 10-gene prognostic model, drug sensitivity analysis was performed. TIDE scores and immune checkpoint expression levels assessed immunotherapy response in high- and low-risk groups. As shown in Fig. 9A, there was no significant difference in TIDE scores between the high- and low-risk groups. Figure 9B shows no significant differences in the expression of eight immune checkpoint genes between risk groups, suggesting a potentially limited response to immune checkpoint inhibitors in high-risk CCA patients. Additionally, we used the predictive model to calculate the sensitivity of high- and low-risk group samples to common antitumor chemotherapeutic and molecular-targeted drugs. Significant differences in sensitivity between the high- and low-risk groups were observed for cisplatin, irinotecan, 5-fluorouracil, and oxaliplatin, while no statistical difference was observed for gemcitabine and paclitaxel (Fig. 9C). It suggests prioritizing high-sensitivity drugs in high-risk patients to reduce toxicity. These results indicate that the 10-gene prognostic model cannot distinguish differences in TIDE scores and immune checkpoint molecule expression between high- and low-risk groups, but it does reveal significant differences in drug sensitivity to cisplatin, irinotecan, 5-fluorouracil, and oxaliplatin.
Fig. 9
Role of Risk Scores in Predicting Immunotherapy and Other Treatments. Note: (A) TIDE scores of CCA patients in high- and low-risk groups. (B) Expression differences of eight immune checkpoints in high- and low-risk CCA patients. (C) Sensitivity analysis of high- and low-risk CCA patients to chemotherapy and small molecule targeted drugs
Comments (0)