
Remember me
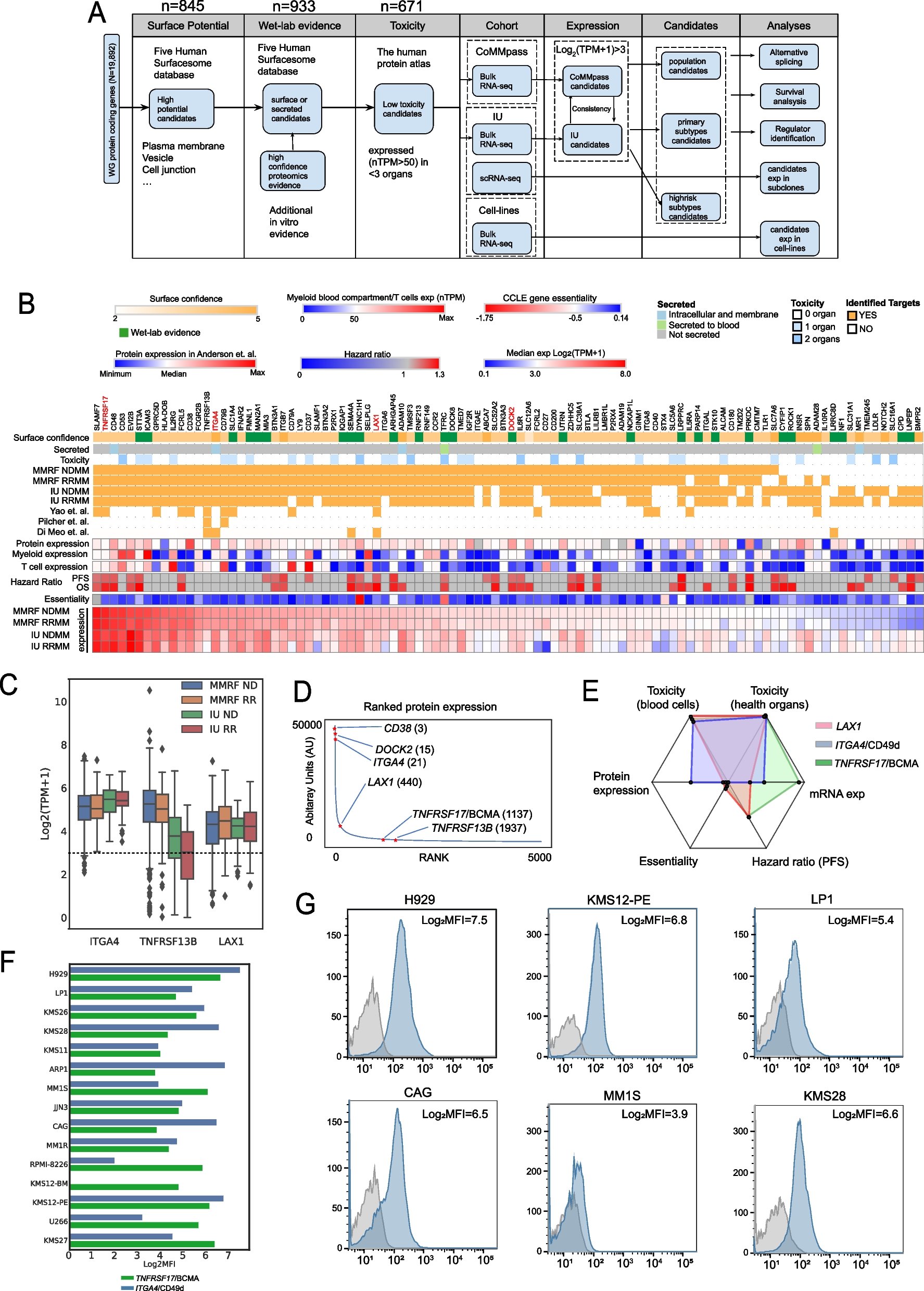




Aging is a complex, multistage process characterized by the progressive decline of organ functionality. Based on cross-organ aging research data published by Stephen R Quake’s team [8], this study systematically analyzed high-throughput RNA sequencing data from 16 mouse organs across 10 time points. Principal component analysis (PCA) revealed significant spatial clustering differences in transcriptomic profiles among different organs (Additional file 1: Fig. S1a), suggesting that each organ may regulate the temporal progression of aging through distinct molecular pathways. To identify key regulatory genes involved in aging, we applied the ImpulseDE2 model to detect differentially expressed genes (DEGs) in each organ (Additional file 1: Fig. S1b-c) and classified them into dynamically similar gene clusters using Mfuzz clustering. These gene clusters were systematically annotated based on eight core aging-related biological processes: cell death, cell division, cell development, immunity, inflammation, hypoxia, DNA repair, and DNA damage. Notably, most organ-specific expression clusters exhibited specific enrichment in these aging-related features, with the lung and kidney showing the strongest associations (Fig. 1a, Additional file 1: Fig. S1d). Among all aging features, cell death and immune function demonstrated the highest levels of association (Additional file 1: Fig. S1e). Different organs exhibited distinct sensitivities to these aging processes. For example, bone tissue clusters were primarily enriched in cell death, cell development, and inflammatory pathways, whereas kidney clusters were predominantly associated with cell death and immune responses. This organ-specific aging signature was further evident at the intra-cluster level. Within the kidney, for instance, Clusters 1 and 4 were specifically enriched in immune and inflammatory pathways, while Cluster 6 was primarily involved in cell division regulation (Fig. 1a, Additional file 1: Fig. S3). These findings indicate that distinct gene modules within the same organ may collaboratively drive aging through functional specialization.
Fig. 1
Identification and functional annotation of aging trend genes. a GO and KEGG annotations of aging trajectory clusters across 16 organs. Left: The Nightingale rose diagram illustrates the number of functional subsets enriched in the organ aging trajectory clusters, with boxes indicating GO terms significantly enriched in aging trajectory clusters. Center: Aging trajectory clusters with enriched GO terms. Right: KEGG pathways linked to aging trajectory clusters; connecting lines colored by organ identity. b Temporal dynamics of aging trajectory clusters in 16 organs (organ labels correspond to panel a). c Statistical summary of aging trend genes, including upregulated, downregulated genes, and organ-specific differentially expressed genes. d Heatmap displaying the temporal proportion of organ samples across different clustering stages. e Heatmap illustrating functional enrichment score at distinct aging stages
Further analysis of dynamic gene cluster patterns revealed a spatiotemporal coupling between gene expression trends and organ physiology. In upward-trending clusters, immune and inflammatory pathways were predominant. For example, Cluster 6 in the heart displayed sustained activation of Th1/Th2 cell differentiation and antigen presentation pathways, indicating an age-related imbalance in T-cell homeostasis [44]. Similarly, Clusters 1 and 4 in the kidney were enriched in chemokine signaling and Th17 cell differentiation pathways, potentially contributing to the development of a chronic inflammatory microenvironment [45]. In contrast, downward-trending clusters were predominantly associated with basal metabolic functions. The progressive silencing of protein digestion and absorption pathways in Cluster 9 of the bone likely reflects a decline in anabolic capacity. Certain clusters exhibited fluctuating expression patterns, such as the intermittent activation of TCA cycle-related genes in the kidney, suggesting transient metabolic remodeling in response to aging-associated stress [46]. Notably, stage-specific high expression in certain clusters correlated with key functional transitions during aging. For example, Cluster 6 in the lung exhibited specific activation of the AMPK/PPAR signaling pathways during late-stage aging, promoting lipid metabolic reprogramming and myofibroblast differentiation [47], thereby exacerbating fibrotic pathology (Fig. 1a,b, Additional file 1: Fig. S4). Cross-organ comparisons revealed that gene clusters with linear temporal changes exhibited conserved aging-related functions, whereas fluctuating clusters were more likely to drive organ-specific processes, whose dynamic features remain challenging to capture.
To systematically decode the continuous molecular evolution during aging, we applied Mann–Kendall (MK) trend analysis to identify aging trending gene (ATG) clusters with significant temporal dependencies. The analysis revealed that ATG clusters were predominantly enriched in cell death and immune-inflammatory pathways, suggesting a cumulative impact of these biological processes throughout aging. In contrast, DNA repair and hypoxia pathways exhibited highly fluctuating activation patterns, likely influenced by transient external stimuli or short-term metabolic demands [48], preventing the establishment of stable temporal trends (Additional file 1: Fig. S1f-g). Genes within ATG clusters, defined as aging trending genes (ATGs), displayed marked heterogeneity across organs. Notably, the small intestine contained the highest number of ATGs (1,389), whereas the pancreas (10 upregulated genes) and skin (129 downregulated genes) exhibited relatively fewer ATGs (Fig. 1c, Additional file 2: Table S2).
Consensus clustering analysis based on ATGs revealed the temporal dynamics of multi-organ aging (Fig. 1d). The results demonstrated that the brain exhibited a unique triphasic aging trajectory, whereas the remaining 15 organs predominantly followed a biphasic transition from a youthful state (State 1) to an aged state (State 2). Notably, organ-specific aging displayed distinct temporal boundaries and heterogeneity. Vulnerable organs, such as the spleen and kidneys, initiated aging programs as early as postnatal month 9, rapidly progressing through state transitions. In contrast, resilient organs, including the skin and bone marrow, retained metabolic activity into old age, exhibiting a relatively slower aging rate (Fig. 1d, Additional file 1: Fig. S2a-e). This differential aging pattern aligns with recent findings on entropy accumulation, where organs such as the spleen and liver show greater entropy increases, suggesting heightened susceptibility to aging-related degenerative changes [49]. Functional annotation analysis further substantiated the molecular basis of organ-specific aging heterogeneity (Fig. 1e). The youthful state exhibited significant enrichment in pathways related to cell cycle regulation and tissue development, whereas the aged state was predominantly characterized by the activation of innate immune responses and inflammatory regulatory pathways. This cross-organ temporal aging pattern unveils a fundamental biological principle: mammalian aging is characterized by a systematic transition from an anabolism-driven developmental program to a survival strategy dominated by inflammatory defense. This discovery not only elucidates the dynamic characteristics of organ-specific aging but, more importantly, establishes a theoretical framework for precisely regulated spatiotemporal aging intervention strategies.
Perturbations of plasma cells and naive-like cells are the main features of organ immune senescenceBuilding upon the pivotal role of immune regulation and programmed cell death in aging, we systematically analyzed the immune microenvironment across multiple organs. Immune infiltration analysis revealed significant age-associated dynamic changes in plasma cells and naive lymphocytes (Fig. 2a, Additional file 1: Fig. S2f-g). Specifically, plasma cells exhibited progressive accumulation in aged organs, whereas naive-like cell populations predominated in young organs. Notably, mesenteric adipose tissue (MAT), white blood cell (WBC), spleen, and lungs exhibited the most pronounced immune remodeling. Quantitative temporal analysis demonstrated a significant positive correlation between plasma cell proportions and aging progression, except in bone marrow (Fig. 2b), whereas CD4 + naive T cells exhibited a significant negative correlation (Additional file 1: Fig. S2h). This trend was further validated using multi-organ bulk-seq datasets from Jonker MJ’s team [25], suggesting cross-organ conservation of this phenomenon (Additional file 1: Fig. S2i). Interaction network analysis further confirmed a strong negative correlation between naive cell proportions and senescence-associated gene expression across five organs, alongside a weak positive correlation with plasma cell accumulation (Fig. 2c). These findings imply that aging promotes immunosenescence through dual mechanisms: suppressing naive cell regeneration while driving plasma cell clonal expansion.
Fig. 2
Dynamic characterization of immune cell type ratios during organ aging. a Immune infiltration landscapes at different stages of organ aging. Top bar graph: Number of organs exhibiting immune cell proportion reconstruction at various aging stages. Left bar graph: Number of immune cell types undergoing significant reconstruction in each organ. Heatmap: Differences in immune cell subset proportions between young (State 1) and aged (State 2) stages, with significance levels indicated (*P < 0.05, **P < 0.01, ***P < 0.001). b The Scatter plot shows the changes in the proportion of plasma cells in different organs over time. c Correlation between the temporal dynamics of immune cell abundances and the expression trajectories of aging trend-associated genes. d UMAP projection showing the annotation of spleen single-cell populations. Right panel: Top: Spatial distribution of AUCell scores for aging-related rising trend gene modules. Bottom: Spatial distribution of AUCell scores for aging-related falling trend gene modules. e Pseudo-time analysis and pie charts illustrating the developmental trajectory of B cell lineages and plasma cells in the spleen, colored by cell types and pseudo-time. f Scatter plots with fitted curves depicting the expression trends of selected marker genes and aging trend genes along the pseudo-time trajectory in the spleen. g Bar graph depicting the ratio of aging to young cells across different immune cell types in the spleen. The data are presented with log2 normalization. h Scatter plot showing changes in cell proportions over time from spleen bulk RNA-seq data deconvoluted using BayesPrism. i Left: scaled cell proportion of different cell types. Each row represents an individual, and each column represents a cell type. Right: averaged cell proportion of different cell types from the two clusters. j The ages of individuals from the three groups. k Schematic showing the cell abundance change of spleen and lung during aging
To further elucidate the role of programmed cell death in aging, we performed ssGSEA enrichment analysis of 12 cell death pathways. Results revealed stage-specific differences in programmed cell death patterns, with the most prominent changes observed in bone marrow and leukocytes, while other organs exhibited relatively modest variations (Additional file 1: Fig. S5a). This divergence may arise from the bone marrow’s hematopoietic centrality, which heightens its sensitivity to cell death, whereas leukocytes depend on programmed death for immune homeostasis [50]. Moreover, inter-organ correlations in programmed cell death patterns were inconsistent, highlighting the complex regulatory networks governing aging-related cellular demise (Additional file 1: Fig. S5b).
Given the significant role of these organs in reshaping immune cell proportions, this study selected MAT, spleen, lung, and subcutaneous adipose tissue (SAT) for further investigation. Single-cell sequencing data were utilized to comprehensively analyze the dynamic changes in their immune microenvironment. A total of 16 cell types were identified in the spleen, 15 in the lung, 9 in the MAT, and 13 in the SAT (Fig. 2d, Additional file 1: Fig. S6a-d, Fig. S7a-d, Fig. S8a-d). By computing the expression scores of aging trend genes via the AUCell algorithm, we observed distinct organ-specific sensitivities to these genes. In the spleen, genes exhibiting an upward aging trend were predominantly expressed in plasma cells, memory B cells, and mature B cells, whereas genes following a downward aging trend were enriched in naive cells and macrophages (Fig. 2d). In the lungs, macrophages and monocytes displayed heightened sensitivity to aging trend gene dynamics (Additional file 1: Fig. S6d-e). Notably, in MAT and SAT, aging trend genes were primarily expressed in stromal cells, particularly endothelial cells (Additional file 1: Fig. S7d-e, Fig. S8d-e), aligning with prior studies indicating that endothelial cells in adipose tissue are highly susceptible to aging and accelerated senescence [51].
To systematically investigate gene expression dynamics and the proportion of senescent cells during aging, we designated the top 10% of cells with extreme expression levels of aging-related genes as characteristic populations exhibiting distinct aging trends, encompassing both upregulated and downregulated subpopulations. Analysis of the spleen microenvironment revealed that plasma cells were predominantly localized within the upregulated aging trend group, while a subset of transitional and activated B cells also exhibited significant activation of aging-related genes (Additional file 1: Fig. S5f). These findings suggest that the B cell lineage may undergo reprogramming of terminal differentiation pathways during aging. To delineate differentiation trajectories, we employed Monocle2 for pseudo-time reconstruction and identified transitional B cells as the developmental origin, diverging into two distinct branches: the Y_107 branch, dominated by plasma cells, and the Y_58 branch, enriched in activated B cells (Fig. 2e,f). The Y_107 branch exhibited significant enrichment in the B cell-mediated immune response pathway, aligning with the established role of plasma cells in terminal differentiation for antibody secretion [49]. Conversely, the Y_58 branch was enriched in cytoplasmic translation and ribosome biogenesis pathways, facilitating protein synthesis to support clonal expansion following antigen stimulation (Additional file 1: Fig. S5h). These findings underscore the potential impact of aging on humoral immune function through modulation of the B cell differentiation program.
Given the spleen’s role in senescent cell clearance, we further examined the influence of natural cellular turnover on our findings. By analyzing the expression levels of gene sets associated with endocytosis and exocytosis, we found that senescence-associated subpopulations exhibited significantly lower phagocytic capacity scores than other cellular subsets (Additional file 1: Fig. S5i-j), indicating that impaired self-clearance in aged cells may contribute to their persistence within the microenvironment. This finding is consistent with the age-related accumulation of plasma cells observed in B-cell differentiation trajectories. Single-cell data from the spleen and lung revealed significant age-related changes in plasma cells and naive-like cells, including a decline in naive-like cells and an increase in plasma cells (Fig. 2g, Additional file 1: Fig. S5g, Fig. S6c). Deconvolution of spleen bulk RNA-seq data using the BayesPrism algorithm, informed by single-cell data, revealed comparable age-associated shifts in cell composition, including a decline in naive CD4 +/CD8 + T cells and an accumulation of plasma cells. Furthermore, we observed a decline in NK T cells and an increase in dendritic cells (DCs), which serve as key regulators of innate immunity [52] and are closely linked to aging (Fig. 2h). Based on a heat map of changes in cell proportions in the spleen, we categorized all volunteers into three age groups and divided each cell type into four groups according to changes in cell proportions. We found that the proportion of cell types in cluster C2 (including plasma cells and DCs) accumulated continuously with increasing age, while the proportion of cell types in cluster C4 (including naive CD4 +/CD8 + and NK T cells) decreased progressively (Fig. 2i–k).
Constructing a novel multi-organ aging assessment (2A) model based on aging trend genesThe remodeling of immune cell proportions across organs demonstrated conserved patterns, with dynamic trajectories closely aligned with the expression of aging trend genes, suggesting the presence of coordinated multi-organ aging mechanisms. Through an analysis of age-associated co-expression patterns of organ-specific aging trend gene sets, we identified significant positive correlations among all organs except bone marrow. The temporal expression similarity of aging trend gene sets across different organs revealed a significant degree of overlap (Fig. 3a). During the identification of conserved cross-organ aging markers, we determined that while organs retained tissue specificity, they also shared core regulatory modules (Fig. 3a). This led us to define 114 global aging trend genes (GATGs), which display consistent trend alterations across at least four organs (Fig. 3b, Additional file 1: S11a). Among these, immune response genes like Ighm, Rbm3, and Igkc increased in 8 organs, while Col3a1, related to collagen synthesis, decreased in 7 organs. Additionally, the GATGs include several known aging marker genes, such as Ccl8 and Tnfrsf1b, and some immune cell markers like Jchain and Cd22 (Fig. 3b, Additional file 2: Table S3). Notably, the kidney, liver, lung, and small intestine accounted for the majority of GATGs, primarily comprising antigen presentation-related genes (Cd74, H2-DMa) and immunoglobulin complex genes (Igkc). The observed inter-organ molecular coordination may originate from shared transcriptional regulatory networks established during embryonic development. Functional enrichment analysis revealed that upregulated genes were predominantly associated with immune-related pathways, including lymphocyte-mediated immune responses, whereas downregulated genes were primarily enriched in protein synthesis pathways, such as collagen fibril organization (Fig. 3c). This finding aligns closely with the classical theory of chronic inflammatory activation and anabolic decline during aging, further reinforcing the hypothesis of coordinated multi-organ aging [45, 53].
Fig. 3
Aging analysis based on organ aging trend gene co-expression patterns. a Top: Heatmap illustrating the expression similarity between organ-specific aging trend genes. Bottom: Bar graph displaying the number of shared aging trend genes among the top six organs. b Global aging trend gene expression across different organs. Outermost circle: Chromosomal locations of global aging trend genes. Inner heatmap: Co-expression patterns of global aging trend genes across various organs, where blue indicates that the gene is an aging trend gene in the corresponding organ. c Functional enrichment analysis of global aging trend genes, categorized into rising trend genes and declining trend genes. d Establishment and verification of the aging assessment model: Left: Flowchart depicting the development of the aging assessment model based on global aging trend genes. Right: Organ-specific aging scores derived from the model. The circular chart visualizes the aging scores of each organ across different stages, while the ridge plot illustrates temporal changes in aging scores for BAT and kidney. e Validation using bulk transcriptome data from young and aged mouse bone tissue (GSE199493) and heart (GSE12480). f Gene set enrichment analysis (GSEA) reveals the enrichment of global aging trend genes in aging states in the Heart cohorts. g The histogram shows the expression levels of classic aging markers Cdkn2a, Ighm, and Ccl8 in bone and heart tissues. h The histogram shows the 2A model scores of the two cohorts
Subsequently, we established a novel multi-organ aging assessment model (2A Model) using global aging trend genes to characterize the overall aging status of each organ (see Methods for details). We calculated the 2A Model score for each organ and conducted a classification evaluation. The results showed that 2A Model scores were consistently higher in aging stages compared to young stages, with scores increasing progressively over time (Fig. 3d, Additional file 1: S9, S10). Sensitivity to the 2A Model score varied among organs: white blood cells (WBC), spleen, and bone marrow exhibited higher scores, consistent with their roles as primary sources of systemic inflammation. In contrast, the brain and lungs had lower 2A Model scores, potentially due to the protective effect of the blood–brain barrier and the specialized regulatory mechanisms of alveolar macrophages. The 2A Model offers a valuable method for tracking the overall aging process and indirectly reflecting age-related changes across different organs.
To assess the universality of global aging trend genes, we systematically integrated mouse heart and bone tissue mRNA-seq datasets spanning young and aged groups (Fig. 3e). Analysis revealed that, compared to the young stage, both organs exhibited upregulation of certain aging markers, such as Ccl8 and Ighm, during aging. However, the classical aging marker Cdkn2a did not exhibit the anticipated upregulation with aging, likely due to its low expression level in bulk RNA-seq (Fig. 3g). To further assess the effectiveness of established aging research tools, we validated two authoritative aging-related gene sets, SenMayo [26] and Aging Atlas [54]. The results indicated that both gene sets effectively characterized age-related changes in heart tissue; however, their discriminative efficiency was markedly reduced in bone tissue, suggesting fundamental differences in tissue-specific aging mechanisms (Additional file 1: S11c). Notably, utilizing the 2A Model scoring system proposed in this study, samples from the aged group exhibited significantly elevated aging indices in both organs, confirming the robustness of GATGs in cross-tissue aging assessment (Fig. 3f–h, Additional file 1: S11b). To investigate the dynamic characteristics of the aging process, we conducted additional validation using a longitudinal time-series dataset. By quantifying the time-dependent correlation strength of organ-specific aging trend genes and GATGs, we observed that both exhibited significant temporal effects. Interestingly, organ-specific aging trend genes demonstrated stronger temporal correlation, suggesting their unique advantage in precisely quantifying tissue-specific aging processes (Additional file 1: Fig. S11d).
The 2A model reveals multi-tissue robustness and single-cell heterogeneity in aging across speciesTo assess the multi-tissue applicability of global aging trend genes, this study utilized human mRNA-seq data from adipose tissue, kidney, pituitary gland, and brain, obtained from the GTEx database. Samples were categorized into young (20–39 years old) and old (60–79 years old) groups based on age, and the recognition efficiency of global aging trend genes in the aging process was analyzed (Fig. 4a–d). The results demonstrated that global aging trend genes, along with the SenMayo gene sets, were significantly enriched across all four tissues (p < 0.001). In contrast, GeneAge exhibited significant enrichment in adipose tissue, kidney and pituitary gland (p < 0.001, 0.05, < 0.001, respectively) but did not reach statistical significance in the brain (p = 0.95). Similarly, Aging Atlas showed significant enrichment in adipose tissue and pituitary gland (p < 0.001) but did not achieve statistical significance in kidney and brain (p = 0.37, 0.94) (Fig. 4a–d, Additional file 1: S11e-h). In further analysis, we calculated the aging scores every 10 years based on the 2A scoring model. The results indicated that the aging scores of these organs showed an upward trend with age, particularly in the kidneys (Fig. 4e–h).
Fig. 4
Global aging trend genes are suitable for aging across species and tissues. Omparative Enrichment Performance of the Global Aging Trend Gene Set vs. GeneAge Database. a Adipose tissue: The global aging trend gene set exhibits significantly stronger aging-associated enrichment (NES = 1.65, FDR < 0.001) compared to the GeneAge database (NES = − 1.62, FDR < 0.001). b Kidney: The global gene set maintains strong enrichment significance (NES = 1.92, FDR < 0.001), whereas GeneAge shows only marginal enrichment (NES = − 1.23, FDR = 0.05). c Pituitary: Both gene sets display significant enrichment, with the global set (NES = 1.87, FDR < 0.001) demonstrating stronger enrichment than GeneAge (NES = 1.59, FDR < 0.001). d Brain: Aging-related enrichment is observed exclusively in the global gene set (NES = 2.48, FDR < 0.001), while GeneAge exhibits no significant association (NES = − 0.82, FDR = 0.95). e–h Ridge plot showing changes in 2A model scores over time for adipose tissue, kidneys, pituitary, and brain, measured in 10-year units. i Schematic illustrating the process of selecting time points for sequencing in studies involving anti-aging treatments. j Histograms showing 2A model scores before and after medication at different time points. k Bar graph depicting the 2A model scores across different lung single-cell types. The first bar represents the score for all cells, followed by scores for specific cell lineage categories
By studying a human cohort [27], we further validated the effectiveness of the 2A scoring model in predicting senescent cell clearance. Neonatal human dermal fibroblasts were cultured to senescence and treated with the anti-aging ATM inhibitor KU-60019. Gene expression changes were measured on days 3 and 8 in both the drug-treated and control groups, each with 3 samples (Fig. 4i). Results showed a significant increase in the 2A model score over time (P = 0.029), and a significant decrease after anti-aging treatment (P = 0.00075) (Fig. 4j). In contrast, GSEA enrichment analysis using individual global aging trend genes failed to effectively identify aging status (Additional file 1: Fig. S11i-j). These findings demonstrate that the 2A scoring model not only assesses overall tissue senescence but also shows significant reductions following senescent cell clearance.
To comprehensively evaluate the performance of the 2A scoring model in assessing aging status at single-cell resolution, we integrated and analyzed a recently published single-cell transcriptomic dataset of mouse lung tissue across multiple time points [55]. Preliminary findings revealed that, with increasing age, globally upregulated aging-associated genes exhibited a marked increase in expression across both tissues and their corresponding cell types, whereas downregulated genes demonstrated consistent attenuation patterns (Additional file 1: Fig. S12). Further benchmarking of the 2A model against two established single-cell aging scoring models, sc-ImmuAging [56] and SCALE [57], demonstrated that at the whole-organ level, the 2A score was significantly and positively correlated with chronological age (R = 0.21, p = 6.9e − 14), and this correlation remained stable across both immune and non-immune cell populations (Fig. 4k). In cell subtype-specific analyses, general capillary endothelial cells and type II alveolar epithelial cells exhibited the strongest age-associated expression trends (R = 0.49 and 0.41, respectively; p < 0.001), suggesting that these may serve as sentinel cell types for monitoring pulmonary aging (Additional file 1: Figure S13d). In model performance comparisons, the 2A model achieved organ-level aging discrimination comparable to that of sc-ImmuAging, and significantly outperformed the SCALE model (Additional file 1: Figure S13a-c). Notably, at the cell subpopulation level, the 2A model effectively captured differential aging trajectories across diverse cell lineages, underscoring its unique advantage and methodological advancement in uncovering cellular heterogeneity during the aging process.
Screening aging-related drugs for multi-organ aging improvementAging is a complex, multifactorial process that results in the gradual decline of cellular, tissue, and organismal function. Our study identified significant time-dependent characteristics in aging trend genes, indicating that anti-aging drugs targeting these genes might produce enduring effects. To capitalize on this, we developed a drug screening model integrating the restart random walk algorithm with iGSEA, tailored to aging trend genes. We used organ-specific and global aging trend genes as inputs, aligning them with corresponding drug targets to construct unique drug-target networks for each organ. By employing adjacency matrices and the random walk algorithm, we calculated drug scores for each organ. Additionally, we sourced gene expression data from drug-treated human cell lines via the Connectivity Map (CMAP) database to derive drug-gene features. Gene set enrichment analysis (GSEA) was then applied using the respective organ-specific aging trend genes to assess the regulatory impact of drugs on aging (see Methods for details) (Fig. 5a, Additional file 2: Table S4).
Fig. 5
Establishment and scoring of aging-related drug identification model. a Schematic diagram illustrating the establishment of an aging-related drug screening model. The model comprises three parts: input objects, data preprocessing, and drug screening walk model. b, c Organ-specific aging trend genes and global aging trend genes were used as input objects to obtain drug rankings, and then the top 10 drugs in each organ and the corresponding targets were screened for network visualization. d Heat map showing the frequency of potential age-related effects of drugs across different organs. e Enrichment analysis and visualization of the targets of Fostamatinib and Ranolazine. f, g Histogram comparing 2A model scores for Panobinostat, Metformin, and the control group
We visualized the top 10 drugs and their target networks for each organ, revealing unique drug-target profiles for each organ and highlighting organ-specific aging characteristics (Fig. 5b,c). Notably, we identified potential anti-aging drugs common to multiple organs, including Fostamatinib, Ranolazine, and Metformin. Fostamatinib, associated with aging in 13 organs, is primarily used for chronic immune thrombocytopenia (ITP) and rheumatoid arthritis (RA) by inhibiting the Syk signaling pathway [58].
In further analysis, we detailed the frequency of drugs across different organs and identified 27 drugs associated with aging in two or more organs, with Fostamatinib and Ranolazine at the forefront (Fig. 5d). Enrichment analysis revealed that Fostamatinib primarily targets pathways related to longevity regulation, circadian rhythm, autophagy, and immunity, while Ranolazine is focused on myocardial contraction, Alzheimer’s disease, and circadian rhythms, all of which are linked to aging (Fig. 5e). We then validated our 2A scoring model using the drug genome, analyzing scores for Panobinostat, which promotes aging, and Metformin, known for its anti-aging effects. Results showed that Panobinostat increased aging GATG scores, while Metformin decreased them, confirming the effectiveness of our 2A scoring model (Fig. 5f,g).
To validate the accuracy of the drug screening model in this study, we integrated aging trend genes into the CMAP database for small-molecule drug prediction and cross-referenced the multi-organ-associated candidates identified by CMAP with our model’s predictions (Additional file 1: S14b). The analysis revealed a strong functional correlation between the CMAP-predicted anti-aging drugs and our model’s screening results. Notably, drugs targeting the mitochondrial respiratory chain complex were identified as potential anti-aging candidates across multiple organs, aligning closely with the mechanisms of metformin and ranolazine, two compounds highlighted by our model [59]. Metformin, for instance, activates the AMPK pathway by inhibiting mitochondrial complex I (NADH dehydrogenase), thereby enhancing insulin sensitivity and regulating glucose metabolism [60]. Similarly, celecoxib, a COX-2 inhibitor, mitigates chronic inflammation by suppressing IKK kinase activity, a mechanism consistent with the regulatory framework of aging-related inflammatory pathways [61]. To further assess the model’s reliability, we conducted organ-specific enrichment analyses on genes exhibiting temporal expression differences under the influence of the identified drugs (Additional file 1: S14c). Notably, the organ-specific drugs predicted by our model effectively distinguished the aging status of the corresponding organs while demonstrating the model’s capability to elucidate organ-specific aging regulatory networks at the molecular level.
Overall, our study underscores the potential of utilizing organ aging trend gene sets to identify and characterize aging-related drug, thereby aiding in the development of novel interventions against aging.
Comments (0)