
Remember me




The study subjects were 2423 individuals of self-declared European ancestry from the TwinsUK cohort [7] (Additional file 1: Table S1). TwinsUK includes about 14,000 monozygotic and dizygotic twin volunteers from all regions across the UK, unselected for any disease and representative of the general population.
Replication cohortThe Qatar Metabolomics Study on Diabetes (QMDiab) is a cross-sectional case–control study of diabetes with 374 participants of Arab, South Asian, and Filipino descent [8, 9] (Additional file 1: Table S1). For 320 samples, joint genetics and glycomics data were available. All study participants were enrolled between February 2012 and June 2012 at the Dermatology Department of Hamad Medical Corporation in Doha, Qatar.
Mass spectrometric glycomicsIn both twinsUK and QMDiab, IgA and IgG glycans were quantified in serum samples using liquid chromatography-mass spectrometry (LC–MS), as previously described [10, 11].
In TwinsUK, blood samples were collected from participants between 1996 and 2016 and were centrifuged at 2000 g for 10 min at room temperature, and instantly stored at − 80 °C before processing. Plasma samples were shipped by courier on dry ice to the Center for Proteomics and Metabolomics, Leiden University Medical Center, and 10 μL of plasma was used for IgG and IgA analysis.
In QMDiab, non-fasting blood samples were collected according with standard protocols as previously described [8]. Briefly, blood for IgA analysis was collected using EDTA tubes, and centrifuged at 2500 g for 10 min at room temperature, plasma was collected, aliquoted and stored at -80 °C until analysis. For each sample, 2 μL and 5 μL of plasma were used for IgG and IgA analysis, respectively.
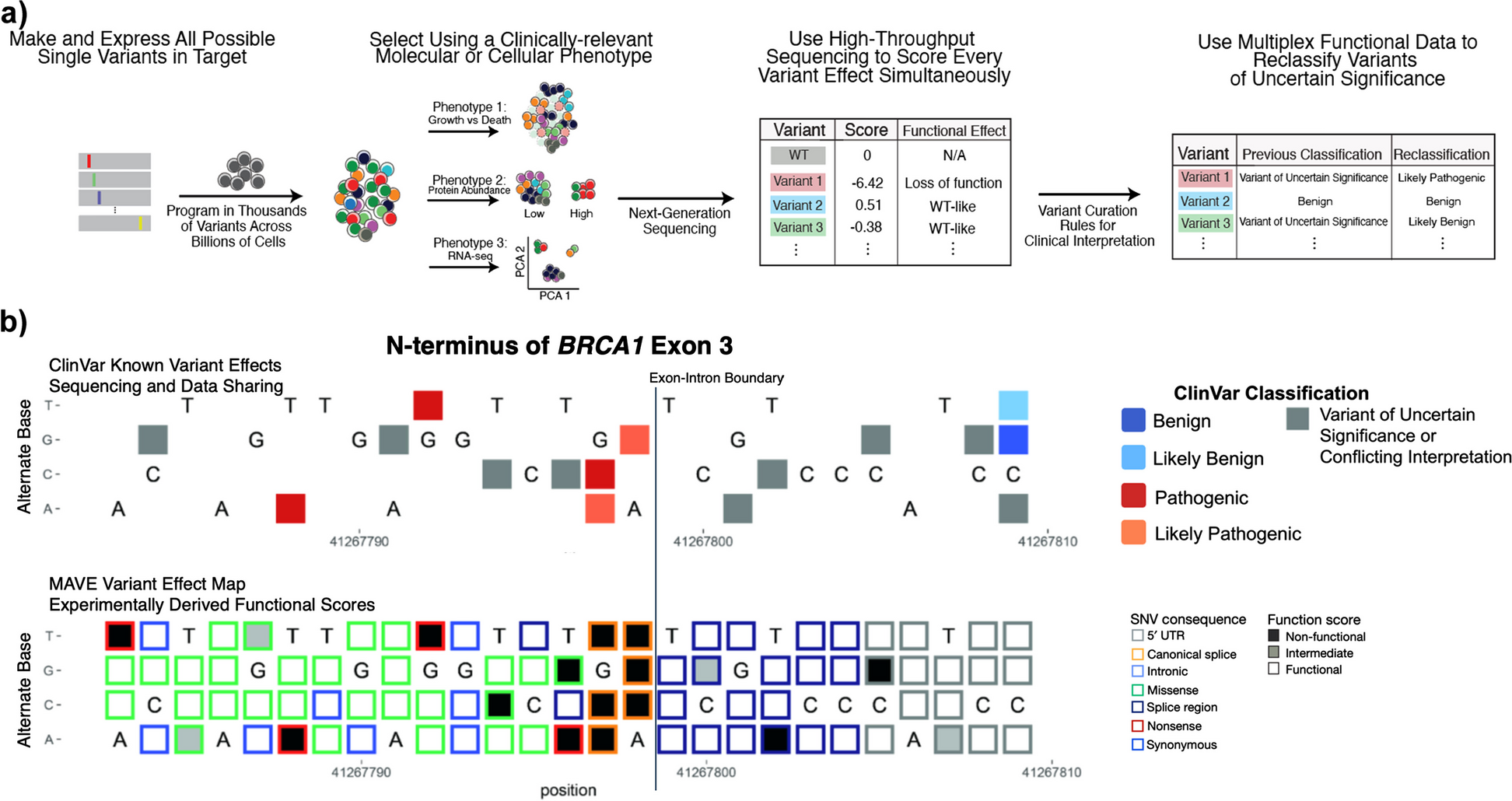
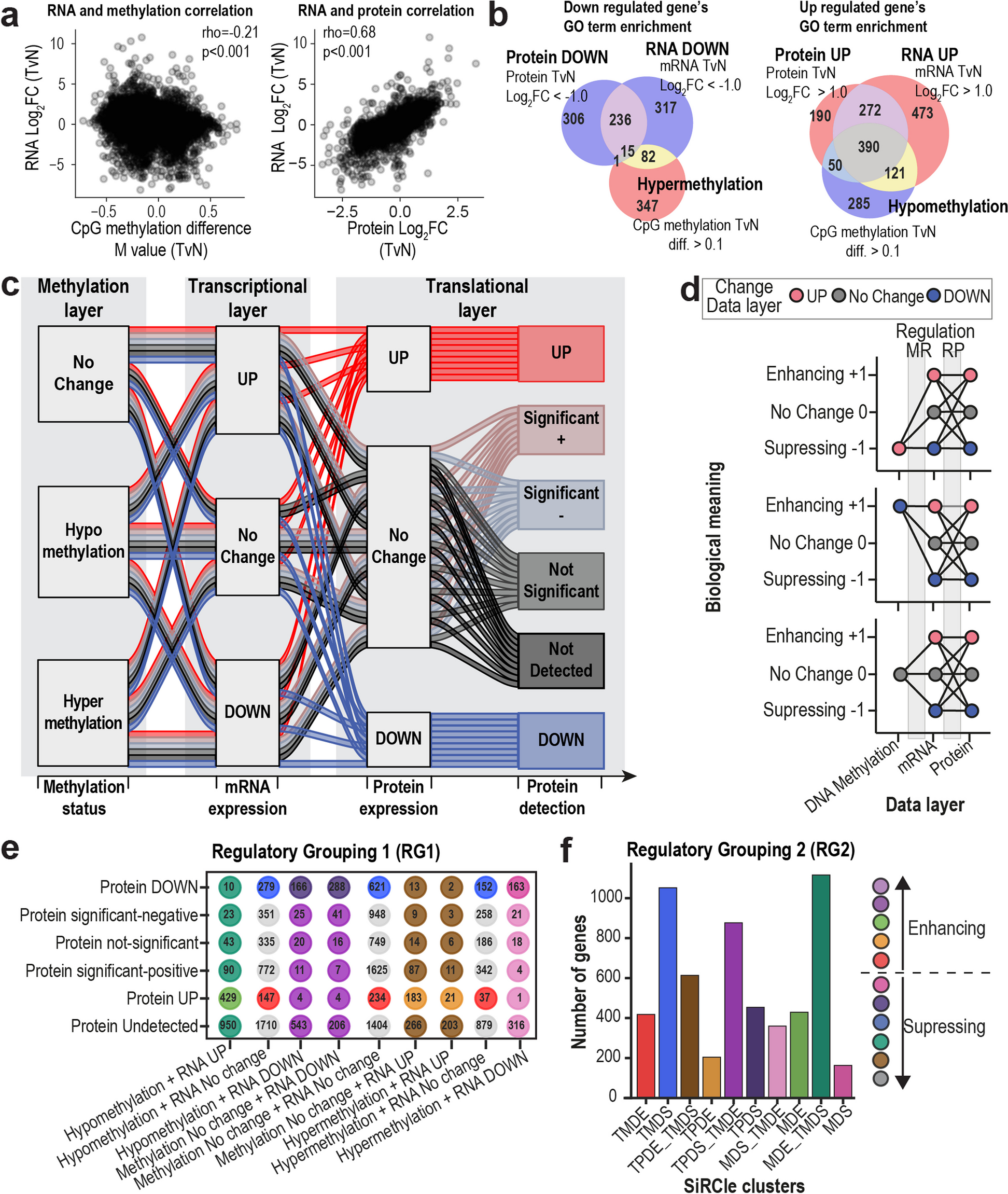
In total, 33 IgA1O-linked glycopeptides, 38 IgA1–2N-linked glycopeptides, and 36 IgG1–4N-linked glycopeptides were retained and quantified. The absolute signal intensities were normalised to the intensity sum of all glycopeptide species sharing the same tryptic peptide sequence, resulting in relative abundances. In this manuscript, IgA1 and IgA2 glycopeptide names are composed of the letter codes of the first three amino acids of the peptide sequence: HYT, LSL, TPL, SES, ENI, and LAG, the last detected in two variants (i.e., as LAGC and LAGY). The peptide name is followed by the glycan composition indicating the number of hexoses (H), N-acetylhexosamines (N), fucoses (F), and sialic acids (Fig. 1a, Additional file 1: Table S2). Glycan structures were assigned on the basis of the tandem mass spectrometric analyses of glycopeptides performed with method development [12] and IgA-released glycan analysis supported by spiking experiments with human plasma standards [13].
Fig. 1
Schematic representation of IgA1 and IgA2 with examples for O- and N-glycan structures. N-glycans are attached to the amino side chain of an asparagine residue (N), while O-glycans are attached to the side chain of serine (S) or threonine (T). Each IgA1 heavy chain contains two N-glycosylation sites (i.e., at N144 and N340), while the hinge region has six O-glycosylation sites (i.e., at T106, T109, S111, S113, T114, and T117), all present on a single tryptic peptide. Both dimeric IgA1 and IgA2 have one additional N-glycosylation site at the J-chain (i.e., at N71). The three-letter code defines the tryptic (glyco-)peptides: HYT for the O-glycosylation at the hinge region of IgA1; LSL for the N-glycosylated sites N144 or N131 on IgA1 or IgA2, respectively; LAG for the N-glycosylated sites N340 or N327 on IgA1 or IgA2, respectively, which were detected with either a terminal tyrosine (LAGY) or as the truncated form (LAGC); SES for the N-glycosylated site N47 on IgA2; TPL for the N-glycosylated site N205 on IgA2; ENI for the N-glycosylated site N71 at the J-chain on IgA2. Glycosylation site numbering is according to Uni-ProtKB. Monosaccharide symbols and example structures of O- and N-glycans and of derived traits are also shown
Structurally similar glycopeptides were summarised into derived traits calculated from their relative intensities (Additional file 1: Table S3), resulting in 7 IgA1O-glycan, 21 IgA1–2N-glycan, and 16 IgG1–4N-glycan derived traits. Because each measured glycopeptide structure carries different types of monosaccharides, derived traits can give a more composite and robust measure of the different glycosylation features. Outliers, defined as measurements deviating more than four standard deviations from the mean of each trait, were removed. To ensure the normality of their distribution, measured glycopeptides and derived traits were quantile normalised.
Association with age and sexThe association of measured glycopeptides and derived traits with age at sample collection and sex was assessed by fitting a linear mixed model in the R statistical framework (lmer function, lme4 package [14] v1.1–31) with plate number and family structure modelled as random effects. We used the approach proposed by Li and Ji [15] to determine the effective number of independent tests to control for the family-wise error rate, resulting in 37 and 24 tests, for IgA and IgG, respectively. Associations were considered significant if the p-value was lower than 0.05/37 = 1.35 × 10−3 and 0.05/24 = 2.08 × 10−3 for IgA and IgG, respectively.
Heritability estimation in TwinsUKWe used the mets R package [16] (v1.2.5) to estimate the contribution of additive genetic (A), shared (C) and individual-specific environment (E) effects on measured glycopeptides and derived traits variations (ACE model), using a classical twin design study. The ACE model was then compared with the most parsimonious AE model, which did not include the effect of the common environment, and the CE and E model, which hypothesised that the trait variability only under environmental control. Models were compared through Akaike’s information criterion (AIC). Quantile-normalised measured glycopeptides and derived traits were corrected for plate number (included as a random effect in a linear mixed model, lmer R function), and passed to mets. Sex and age at the sample collection were included as covariates. Four pairs of monozygotic twins and three pairs of dizygotic twins were removed because the twins were either adopted or reared apart.
IgA and IgG intra-class correlationsFor both isotypes, intra-class (i.e., IgA-IgA and IgG-IgG) pairwise correlation between both measured glycopeptides and derived traits was estimated using Pearson’s product–moment correlation on pre-processed glycan abundances. More in detail, quantile-normalised measured glycopeptides and derived traits were adjusted for age, sex (fixed effects), plate number, and family structure (random effects) using a linear mixed model (lmer R function), and the correlations were computed on the residuals. Associations were considered significant if the absolute correlation coefficient was greater than 0.25 and the p-value was lower than 0.05/(N × (N − 1)/2), where the denominator corresponds to the number of elements of a lower triangular matrix with size N × N, and N corresponds to the effective number of independent tests as described above. The value of N was 37 and 24, for IgA and IgG, respectively.
IgA glycan ratiosRatios were calculated using untransformed relative frequencies of pairs of measured glycopeptides expressed on the same peptide, after outliers were removed. For N-glycans, we selected IgA and IgG measured glycopeptides representing the product–substrate of known sialylation and galactosylation reactions in the Ig N-glycosylation pathway [17]. For O-glycans, reactions of N-acetylgalactosaminylation, sialylation, and galactosylation were estimated based on the ratios of pairs of measured O-glycopeptides which differed for a single GalNAc, sialic acid and galactose residue, respectively. Overall, 21 multi-step enzymatic conversions were identified, involving at least two enzymatic transformations of the same underlying glycan structure, as exemplified below:
$$TPL\_H5N5F1S0\;\overrightarrow\;TPL\_H5N5F1S1\overrightarrow\;TPL\_H5N5F1S2$$
Differences in the distribution of ratios representing two consecutive glycosylation reactions were assessed using the Wilcoxon test, and we considered significant p-values lower than 0.05/31 = 2.38 × 10−3, where 31 corresponds to the number of comparisons performed.
IgA-IgG shared heritabilityPairwise additive genetic (ρG) and environmental (ρE) correlations among IgA-IgG derived traits were estimated through bivariate maximum likelihood-based variance decomposition in SOLAR-Eclipse [18] (v8.1.1; http://www.solar-eclipse-genetics.org/). Age and sex were included as covariates. SOLAR-Eclipse was also used to estimate the phenotypic correlations (ρP) so that the same proportion of variance associated with covariates is removed for phenotypic and genetic correlation estimates. Likelihood ratio tests were used to assess whether ρP, ρG, and ρE were significantly different from zero. Phenotypic correlations were considered significant if the absolute correlation coefficient was larger than 0.25 and the associated p-value was lower than 0.05/(37 × 24) = 5.63 × 10−5, where 37 and 24 correspond, respectively, to the effective number of independent IgA and IgG, estimated as described above.
For pairs of correlated IgA-IgG glycan traits, we estimated the genetic (COVG) and environmental (COVE) covariances based on the following formulas:
$$}_ (x,y) =_(x,y) \times \sqrt_^}\times \sqrt_^}$$
$$}_ (x,y) =_(x,y) \times \sqrt_^}\times \sqrt_^}$$
where h2x and h2y are the heritabilities (h2) of traits x and y, as estimated by SOLAR-Eclipse, and \(_(x,y)\) and \(_(x,y)\) are the genetic and environmental correlations, respectively. Genetic/environmental correlation coefficients were constrained to zero if the associated p-value was larger than 0.05, so that the resulting covariance is zero. Then, the proportion of traits phenotypic correlation explained by genetics (i.e., shared heritability) was estimated based on the following equation:
$$_^ =\frac}_ (x,y)}}_ (x,y) + }_ (x,y)}$$
which assumes that the total phenotypic covariance is given by the sum of the genetic and environmental covariances COVG and COVE, respectively.
GenotypingIn TwinsUK, microarray genotyping was conducted using a combination of Illumina arrays (HumanHap300, HumanHap610, 1 M-Duo and 1.2 M-Duo 1 M) and imputation was performed using the Michigan Imputation Server [19] using haplotype information from the Haplotype Reference Consortium (HRC) panel [20] (r1.1). Genotypes were available for 2371 individuals with glycomics data. A total of 5,411,460 single nucleotide polymorphisms (SNPs) meeting the following conditions were included in our genome-wide association study: call rate ≥ 95%, minor allele frequency (MAF) > 5%, and imputation score > 0.4.
In QMDiab, genotyping was carried out using Illumina Omni array 2.5 (v8). Standard quality control of genotyped data was applied, with SNPs filtered by sample call rate > 98%; SNP call rate > 98%, and Hardy–Weinberg equilibrium (HWE) P < 1 × 10−6. Imputation was done using SHAPEIT software [21] with 1000G phase 3 version 5 and mapped to the GRCh37 human genome build. Imputed SNPs were filtered by imputation quality > 0.7.
Genome-wide association study (GWAS) discovery stepTo take into account the non-independence of the twin data, the association with IgA and IgG measured glycopeptides and derived traits in the TwinsUK cohort was performed using GEMMA [22] (v0.98.1), assuming an additive genetic model and including sex, age at the sample collection, the first five principal components assessed on the genomic data, and plate number as covariates. Associations were considered significant and taken forward for replication if their discovery p-value was lower than 5 × 10−8/37 = 1.35 × 10−9 and 5 × 10−8/24 = 2.08 × 10−9, for IgA and IgG, respectively.
Identification of independent signals within lociWe used a stepwise procedure to identify independent signals within the loci identified in the discovery cohort. For each locus, we fitted a new regression model, where the top-associated genome-wide significant SNP was included as a covariate (i.e., conditional model). Genome-wide significant (P < 5 × 10−8) SNP resulting from the conditional model was considered as an independent signal and was included in the covariate set of a new conditional model. We stopped the stepwise procedure when we could not identify any additional genome-wide significant SNPs. Conditional models were built using GEMMA [22] (v0.98.1).
Replication stepWe attempted replication of independent signals for IgA measured glycopeptides and derived traits using data from the QMDiab cohort. Replication was evaluated at the lead SNP of each locus, or at a tag SNP in high linkage disequilibrium (R2 ≥ 0.8, distance ≤ 500 kb) with any genome-wide significant SNP within the locus. The linkage disequilibrium (LD) structure was assessed with LDlink [23] (v3.6) using the available GBR population from 1000 genomes project phase 3. Genetic association was conducted using a linear regression model adjusting for age, sex, diabetes status, and the first three genetic principal components, and was considered replicated if the direction of the effects was concordant between the two cohorts, and if the p-value was lower than 0.05/72 = 6.94 × 10−3, where 72 is the number of IgA measured glycopeptides and derived traits-genomic locus pair identified in the discovery step.
To assess the replicability of loci reported by previous GWAS for serum IgG N-glycans, we retrieved the reported lead SNPs for the 27 independent genome-wide significant loci identified by the largest (n = 8,090) GWAS published to date on circulating IgG glycome [2]. We considered an association replicated in TwinsUK if the p-value of the association for any IgG measured glycopeptides and derived traits at the lead SNP of each locus was lower than 0.05/(27 × 24) = 7.72 × 10−5, where 27 is the number of loci tested and 24 is the effective number of IgG measured glycopeptides and derived traits tested.
Identification of known lociWe interrogated the NHGRI-EBI GWAS Catalog [24] (release 2023–02-03, association P < 5 × 10−8) to identify previously reported associations in coincidence or in strong linkage disequilibrium with those identified and replicated in our study. Additionally, we queried our results to identify associations in coincidence or in strong linkage disequilibrium with 25 genome-wide significant loci reported by a large IgAN meta-analysis [25], and passing a Bonferroni-derived threshold of 0.05/25 = 0.002. Linkage disequilibrium was assessed using LDlink [23] with the parameters listed above.
Variants annotationLocus-gene mapping was performed using OpenTarget [26] (https://genetics.opentargets.org/, release 22.10, October 2022). OpenTarget used the “locus-to-gene” (L2G) model to prioritise likely causal genes. An L2G score is derived for a given SNP–gene pair by aggregating evidence from different mapping strategies, including physical distance from the transcription start site of nearby genes, (sQTL) and expression (eQTL) quantitative trait loci colocalization, and chromatin interaction mapping, as well as in silico predictions of SNP functional consequence.
Shared genetic effectFor each pair of derived traits for which a replicated genome-wide significant signal was identified, we applied a Bayesian bivariate analysis, as implemented in the GWAS-PW software [27] to investigate the presence of a shared genetic effect in the TwinsUK cohort. Briefly, GWAS-PW estimates the posterior probability of quasi-independent genomic regions to include a genetic variant which (i) associated only with the first derived trait (model 1), (ii) associated only with the second derived trait (model 2), (iii) associated with both derived traits (model 3) or (iv) that the genomic block includes two genetic variants, associating independently with each of the two derived traits (model 4). We defined regions for which a genome-wide significant signal was identified for at least one of the two traits and showed a posterior probability larger than 0.9 for model 3 or model 4 as characterised by an underlying shared genetic effect or by colocalization of effects, respectively.
Quasi-independent regions (n = 1703) were pre-determined by estimating LD blocks in European populations [16]. Since our GWASs were performed on overlapping sets of individuals, we estimated the expected correlation in the effect sizes from the GWAS summary statistics using fgwas [28], as suggested in [27], and used this correction factor in the GWAS-PW modelling.
SNP-based heritabilityFor a given SNPi, we calculated the proportion of explained phenotypic variance in TwinsUK as:
$$_} = 2\times }_}\times }_}\times _}}^$$
where βi is the estimated effect of SNPi in univariate analysis, and pi and qi are the minor and major allele frequencies of SNPi respectively, as estimated in TwinsUK. For measured glycopeptides and derived traits that had more than one independent genome-wide significant association, we estimated the total SNP-based variance explained as the sum of the proportion of variance explained by each of the top independent SNPs.
GWAS signals contribution to IgA-IgG shared heritabilityTo evaluate the contribution of GWAS signals to IgG-IgA glycome shared heritability, we estimated pairwise IgA-IgG derived traits additive genetic covariance upon conditioning on the lead SNPs in TwinsUK. More in detail, for each pair tested, derived traits were independently adjusted for age, sex, plate number, and lead SNPs at genome-wide-significant loci associated with any of the two traits (lmer R function). Standardised residuals were then passed to SOLAR-Eclipse [18] (v8.1.1) to estimate the conditional phenotypic and genetic correlations. The additive genetic covariance was estimated based on the following formula:
$$}_\textit(x,y)=A_x\times A_y\times\rho G_$$
where Ax and Ay are the square roots of the heritabilities (h2) of traits x and y, and ρGxy is the additive genetic correlation.
Comments (0)