
Remember me
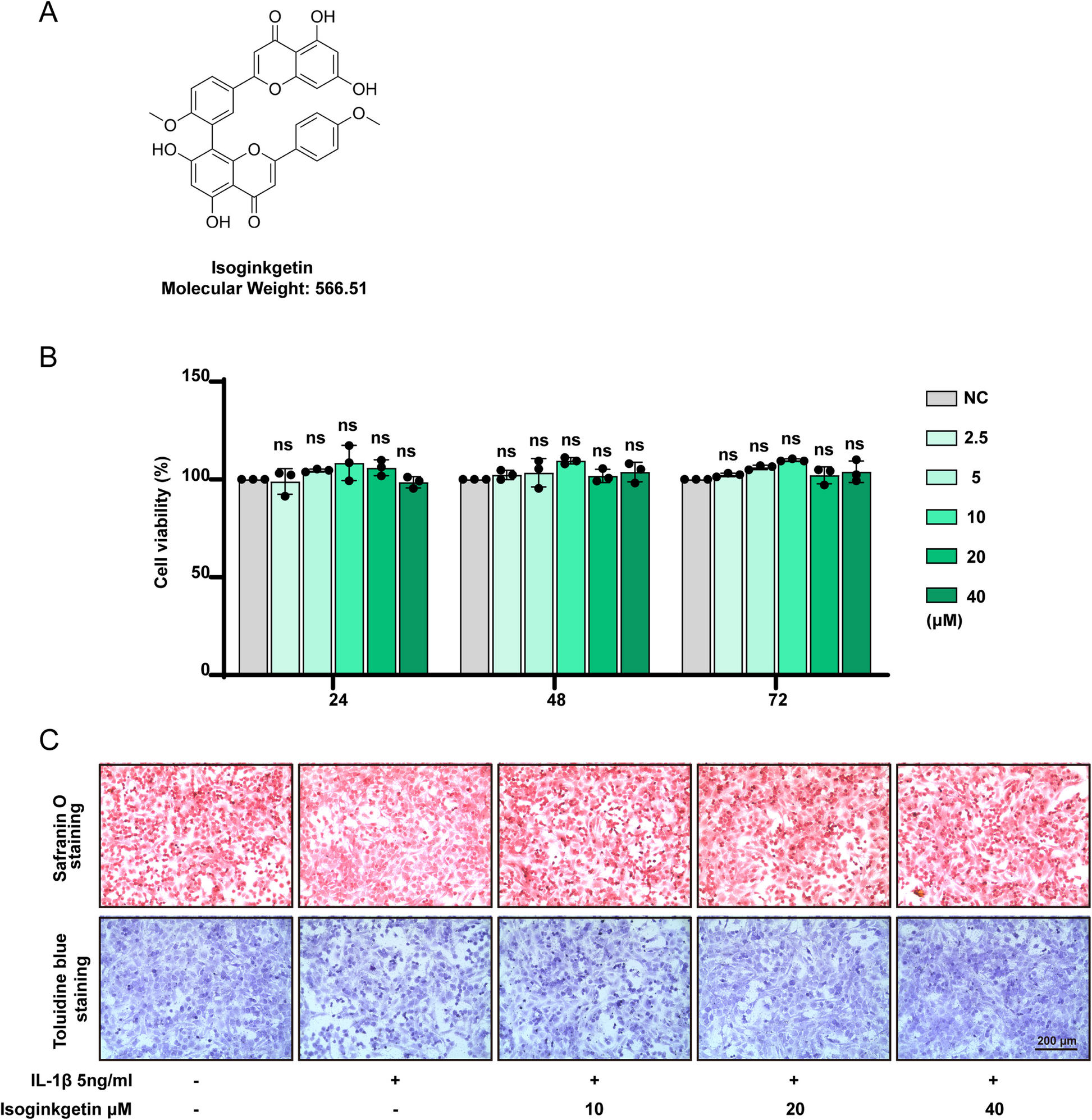





Dehydrocostus lactone (DHL) with a purity of 98% (catalog number: B21026; HPLC analysis) was obtained from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). The molecular formula of DHL is shown in the figure on the right. Simvastatin (catalog number: S831014; 98% purity, HPLC analysis) was purchased from Shanghai Macklin Biochemical Technology Co., Ltd (Shanghai, China).
Before the treatment, DHL and Simvastatin were dissolved in a mixed vehicle solution composed of 10% dimethyl sulfoxide (DMSO), 40% PEG300, 5% Tween-80, and 45% saline to obtain the stock concentrations of 20 mg/mL and 60 mg/mL, respectively.
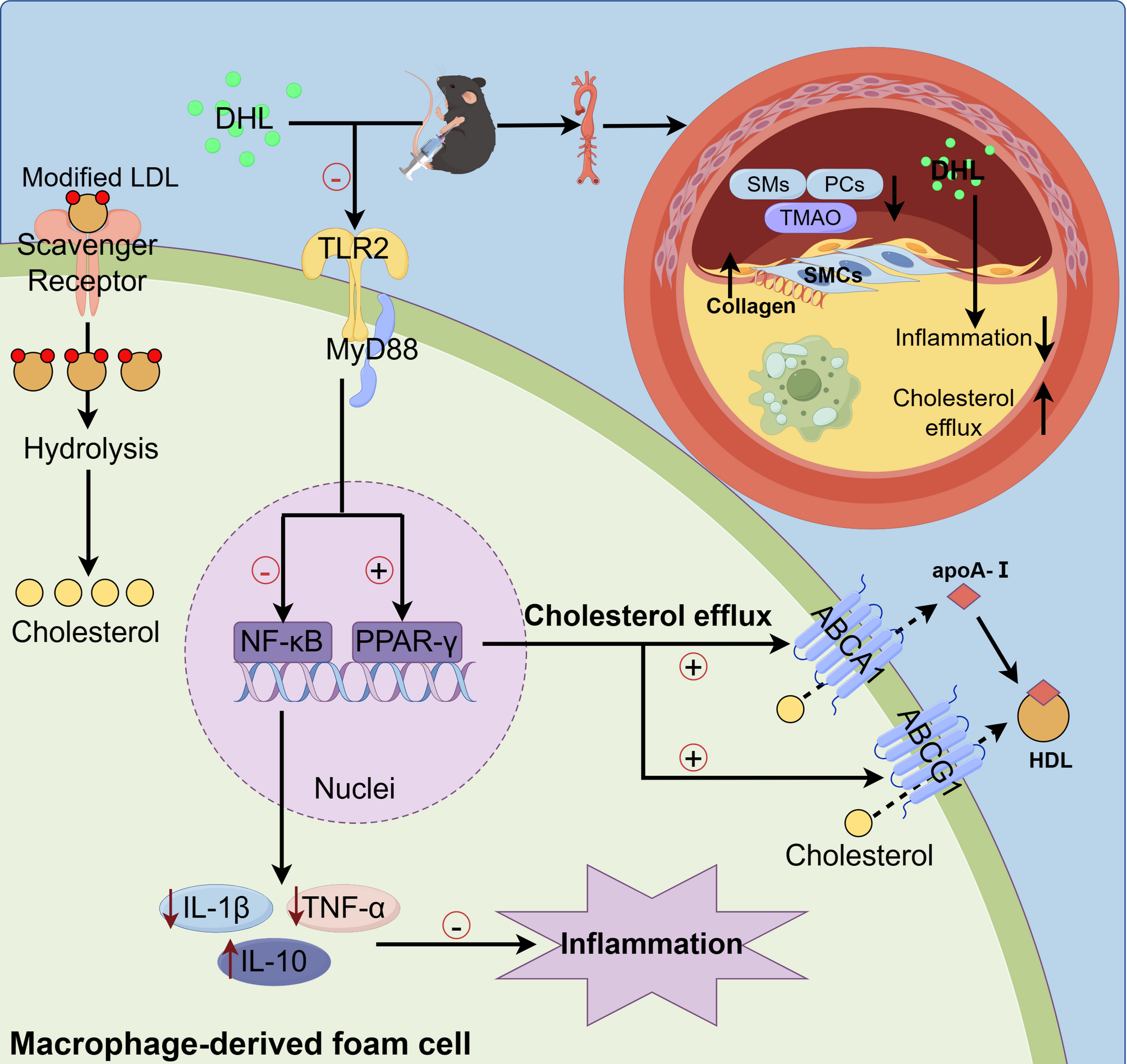
Animals and treatmentWild-type (WT) mice and ApoE−/− mice (6–8 weeks old, weighing 20–25 g) were obtained from GemPharmatech Co., Ltd. (Strain ID: T001458, Nanjing, China). All animals were housed under controlled conditions, maintaining a temperature of 24 ± 2℃, a humidity level of 50 ± 5%, and a standard 12-hour light/dark cycle. The animal facility ensured a pathogen-free environment. A total of 48 mice (8 WT mice and 40 ApoE−/− mice) were randomized into the control group (WT mice fed a standard chow diet), the AS group (ApoE−/− mice fed a high-fat diet consisting of 89% basal diet, 10% lard oil, and 1% cholesterol) with vehicle solution treatment, AS with low concentration of DHL (10 mg/kg/day, L-DHL group) treatment, AS with a moderate concentration of DHL (20 mg/kg/day, M-DHL group) treatment, AS with a high concentration of DHL (40 mg/kg/day, H-DHL group) treatment, and the simvastatin group (6 mg/kg/day). Each group comprised 8 mice. The mice were fed a high-fat diet for 8 weeks to induce atherosclerosis, followed by a 10-week treatment with DHL and simvastatin via intraperitoneal injection at every 2 days (3:00 p.m.). The injection dosage was calculated according to the drug concentration and individual animal weight. Mice were provided with ad libitum access to food and water throughout the experiment.
Blood lipid determinationApproximately 1 mL of whole blood samples was obtained from the angular vein of anesthetized mice using pentobarbital. Serum samples were obtained by centrifugating the whole blood at 1790 × g for 10 min at room temperature. Then, the serum samples were stored in microtubes at −80 °C. The serum levels of triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) were measured using commercially available kits purchased from Applygen Technologies Inc. (E1003, E1005, E1017, and E1018; Beijing, China).
Aortic plaques assessmentMice were euthanized by cervical dislocation in accordance with ethical guidelines. The entire aorta was carefully dissected, and the adipose tissue surrounding the arterial structure was thoroughly removed. Then, they were stored in a 4% paraformaldehyde solution in PBS at room temperature for 24 h. The oil red O staining was performed to assess the plaque level of the whole aorta. In brief, the aorta was longitudinally cut along the vessel wall and soaked in oil red O solution (G1016, Servicebio, Wuhan, China) for 60 min. Subsequently, it was differentiated using 75% ethanol until the fatty plaques in the lumen appeared orange or bright red, while the other parts became nearly colorless. Finally, the aortas were rinsed twice with distilled water.
Analysis of atherosclerotic plaques in aortic rootThe aortas were fixed in 4% paraformaldehyde in PBS for 24 h at room temperature. Following fixation, the aortas were dehydrated in a 30% sucrose solution and subsequently embedded in an optimal cutting temperature compound. Each sample was then sectioned into 5 slices, with each slice containing 3 sections of 8 μm thickness from aortic root. Hematoxylin and eosin (H&E) staining was performed on the sections, which were later sealed with neutral resin for analysis of arterial morphological changes. Lipid content in the plaques was visualized analysis using oil red O staining, while Masson staining was employed to assess the collagen content. The level of smooth muscle cells was determined by immunohistochemical staining of alpha-smooth muscle actin (α-SMA) using a rabbit polyclonal antibody (pAb, GB111364, Servicebio, Wuhan, China). For analysis of macrophage polarization in the plaques, immunofluorescence staining was carried out using anti-CD68 rabbit pAb (GB113109, Servicebio, Wuhan, China) and anti-CD163 pAb (GB113751, Servicebio, Wuhan, China). Finally, the CaseViewer software was utilized to calculate the positive area ratio of the slides (Yang et al. 2025).
Determination of cytokine levels in serum and foam cellsThe serum concentrations of interleukin (IL)−1β, IL-10, and tumor necrosis factor-α (TNF-α) were determined using ELISA kits (EMC001bQT, EMC005, and EMC102a; NeoBioscience Technology Co., Ltd., Shenzhen, China).
The levels of IL-1β, IL-10, and TNF-α in the culture supernatant were quantified using ELISA kits. Detection of absorbance at 450 nm was performed using a microplate reader. Concentrations of TNF-α, IL-1β, and IL-10 in each group were calculated using the standard curve.
Serum metabolomics assayEighteen serum samples collected from WT group (6 samples), AS group (6 samples), and H-DHL group (6 samples) were submitted for metabolomics analysis. Serum metabolomic profiling was conducted in collaboration with Gene Denovo Biotechnology Co., Ltd. (Guangzhou, China), which encompassed mass spectrometry analyses and/or bioinformatics analysis. Multivariate statistical analysis was performed following the processing and annotation of the original LC-MS data. Discrimination of metabolites between the two sample classes was achieved by applying a statistically significant threshold, notably the Variable Importance in Projection (VIP) value (VIP > 1), which was further validated through Student’s t-test analysis (P ≤ 0.05). For the comparison of groups, the Orthogonal Projection to Latent Structures-Discriminant Analysis (OPLS-DA) was employed utilizing R package models. Additionally, to explore alternative metabolic pathways, differential metabolites underwent grouping and enrichment analysis using MetaboAnalyst 5.0 in conjunction with the KEGG database.
Quantitative RT-PCR analysisTotal RNA samples were isolated from the aorta and macrophages using Trizol reagent (15596026, Invitrogen™, Carlsbad, CA, USA). Subsequently, a reverse transcription was performed on 500 ng of total RNA in a 10 µL system to synthesize cDNA, employing the PrimeScript™ RT reagent Kit with gDNA Eraser (RR047 A, Takara, Beijing, China). Quantitative PCR (qPCR) was conducted to analyze the expressions of target genes, utilizing SYBR Green I dye under the following conditions: 95 °C for 30 s, 95 °C for 5 s, 60 °C for 34 s, with 40 cycles, followed by 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s for one cycle. The primer sequences used for the target genes, including TLR2, TLR4, myeloid differentiation factor 88 (MyD88), nuclear factor kappa B (NF-κB), PPAR-γ, LXR, ABCA1, ABCG1, IL-1β, IL-10, TNF-α, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are listed in Table 1. Relative mRNA expressions of the target genes compared to GAPDH were calculated utilizing the 2−∆∆CT method.
Table 1 Primer sequences used for real-time RT-qPCRImmunohistochemistryImmunohistochemistry was employed to assess the expression of TLR2 (GB11518, Servicebio, Wuhan, China) and MyD88 (GB111554, Servicebio Wuhan, China) in the aortic root. The slides were prepared by paraffin-embedding, slicing, and subsequent deparaffinization. Antigen retrieval was performed by heating the slides in a microwave oven for 20 min in a 0.1 mol/L sodium citrate solution, followed by treatment with 1% hydrogen peroxide to eliminate endogenous peroxidase activity. The slides were then blocked with 2% goat serum for 1 h. Primary antibodies were applied at a dilution of 1:200 in goat serum and incubated overnight at 4℃. This was followed by incubation with horseradish peroxidase conjugated goat anti-rabbit immunoglobulin G (1:500; A0208, Beyotime Biotechnology, Shanghai, China) for 2 h at room temperature. Nuclei were counterstained with hematoxylin for 5 min. The slides were mounted with coverslips and visualized under a light microscope (magnification, ×500; Nikon Corporation). ImageJ 1.45 software (US National Institutes of Health, Bethesda, MD, USA) was utilized for analysis.
Macrophage’s culture and foam cell formationMouse RAW264.7 cells (CL-0190, Pricella Biotechnology Co., Ltd, Wuhan, China) and human monocytic THP-1 cells (CL-0233, Pricella Biotechnology Co., Ltd, Wuhan, China), were cultured in a 37 °C incubator with 5% CO2. RAW264.7 cells were maintained in DMEM high-glucose media, while THP-1 cells were maintained in RPMI 1640 high-glucose media, both supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (100 units/mL penicillin and 100 µg/mL streptomycin). To induce the differentiation of THP-1 monocytes into macrophages, 160 nmol/L PMA was utilized. The formation of macrophage-derived foam cells was achieved by incubating the cells with a serum-free medium (DMEM or RPMI 1640) containing 0.2% bovine serum albumin (BSA) and 50 µg/mL acetylated-low density lipoprotein (ac-LDL; JK-011; Anhui Jingke Biotechnology Co., LTD, Anhui, China) for 48 h.
Cholesterol efflux assayTo determine cholesterol efflux, RAW264.7 macrophages (1 × 104 cells/well) and THP-1 macrophages (1.5 × 104 cells/well) were seeded into 24-well plates. The cells were then incubated with a serum-free medium consisting of 0.2% BSA, 2 µg/mL NBD-cholesterol (810250P, Avanti Research™, Alabama, United States), and 50 µg/mL ac-LDL for 48 h. Following this, the cells were incubated with a serum-free DMEM (or RPMI 1640) consisting of 0.2% BSA for 12 h to balance the intracellular lipid. Finally, the cells were incubated with varying concentrations (0, 1, 5, and 10 µmol/L) of DHL in serum and phenol red-free DMEM (or RPMI 1640) medium for 12 h to induce cholesterol efflux. The levels of NBD-cholesterol in cells and medium were measured using a microplate reader by detecting excitation light at a wavelength of 465 nm and emission light at 535 nm. Cholesterol efflux (%) was calculated by dividing the fluorescence intensity in the efflux media by the total fluorescence intensity (media plus cells) and multiplying the result by 100%.
To quantify cellular cholesterol levels, foam cells derived from macrophages were subjected to a 12-hour treatment with or without 5 µmol/L DHL. TC level was measured using an assay kit (E1015, Applygen Technologies, Beijing, China). Protein concentration in the same lysate was determined using the Enhanced BCA Protein Assay Kit (P0009, Beyotime Biotechnology, Shanghai, China). Total cellular cholesterol was reported as nmol/mg protein, with results presented as the mean ± standard deviation of three independent experiments. Oil red O staining was employed to assess lipid droplet levels in the cells, and images were captured using an inverted light microscope (Axio Vert. A1, Carl Zeiss, Jena, Germany).
Western blot analysisTo validate the lipid-lowering and anti-inflammatory properties of DHL, the cells were incubated with 0, 1, 5, and 10 µmol/L of DHL in the presence or absence of TLR2 agonist Pam3 CSK4 (tlrl-pm2 s-1, Invivogen™, Carlsbad, CA, USA) and the TLR2 inhibitor C29 (HY-100461, MedChem Express, Shanghai, China).
Total protein samples were collected by extracting cells in RIPA lysis buffer with a protease inhibitor (P1006, Beyotime Biotechnology, Shanghai, China). The resulting mixture was then centrifuged at 12,000 × g for 10 min at 4 ℃. Subsequently, 30 µg of total proteins per lane were separated using 8%, 10%, or 12% SDS-PAGE, depending on the size of the target proteins. These proteins were then electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes in a trans-buffer at 100 V for 1 or 2 h. For blocking, PVDF membranes were treated with either 5% skimmed milk or 5% bovine serum albumin (for phosphorylated proteins) in TBST buffer (20 mM Tris-HCl, pH 7.5, 150 mM sodium chloride, and 0.05% Tween 20) for 2 h. After three washes with TBST buffer for 5 min each, the membranes were incubated overnight with primary antibodies against MyD88 (4283 S, Cell Signaling Technology, Shanghai, China), NF-κB (8242 S, Cell Signaling Technology, Shanghai, China), PPAR-γ (ab272718, Abcam, Shanghai, China), LXR (sc-377260, Santa cruz, Texas, USA), ABCA1 (ab66217, Abcam, Shanghai, China), ABCG1 (MA5-35185, Thermo, Shanghai, China), and GAPDH (2118 S, Cell Signaling Technology, Shanghai, China). Subsequently, the membranes were incubated with anti-rabbit (A0208, Beyotie Biotechnology, Shanghai, China) or anti-mouse (A0216, Beyotime Biotechnology, Shanghai, China) secondary antibodies conjugated to horseradish peroxidase (1:2000) at room temperature for 2 h. Protein bands were detected using enhanced chemiluminescence and quantified with Fiji software (W. Rasband, NIH, USA), with GAPDH as the normalizer.
Molecular dockingTo analyze the binding affinities and modes of interaction between DHL and TLR2, AutodockVina 1.2.2, a silico protein-ligand docking software was employed (Morris et al. 2008; Xu et al. 2024). The molecular structure of DHL (PubChem CID: 73174) was retrieved from PubChem Compound (Wang et al. 2017a, b) (https://pubchem.ncbi.nlm.nih.gov/). The 3D coordinate of TLR2 (PDB ID: 6 NIG; resolution, 2.35 Å) was downloaded from the PDB (http://www.rcsb.org/pdb/home/home.do). For docking analysis, all protein and molecular files were converted into PDBQT format with all water molecules excluded and polar hydrogen atoms were added. The grid box was centered to cover the domain of each protein and allow for unrestricted molecular movement. The grid box was set to 30 Å × 30 Å × 30 Å, and grid point distance was 0.05 nm. Molecular docking studies were performed by Autodock Vina 1.2.2 (http://autodock.scripps.edu/).
Statistical analysisQuantitative data are reported as mean ± SD from the independent experiments. Statistical analysis was performed using Prism 9 (GraphPad, San Diego, CA, USA) software. The multiple comparisons were conducted by one-way ANOVA with Tukey’s test. Significance was defined as P < 0.05.
Comments (0)