Preparation of HA-A. platensis and bacterial culture
In this investigation, a 40-gram batch of A. platensis powder was utilized and subjected to homogenization using a solvent mixture composed of ethanol and water in a 60:40 ratio (v/v), with a total volume of 200 mL. The homogenization procedure involved stirring the mixture for a duration of two hours. Following the homogenization process, the resulting supernatants were filtered and meticulously collected. The solvents were subsequently concentrated using a rotary vacuum evaporator and freeze-dried to yield the final freeze-dried HA-A. platensis. The freeze-dried samples were stored at a temperature of 4 °C for subsequent analysis [10].
The strain L. helveticus IBRC-M 11,312, graciously provided by Dr. Baradaran Ghavami from the Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center at the Research Institute, was cultured in Man, Rogosa, and Sharpe (MRS) broth at 37 °C for 48 h until it reached the late exponential phase with a density of 107 colony-forming units (CFU)/mL. In this study, L. helveticus was employed in three different forms: live bacterial cells (LBC), heat-killed bacterial culture (HKC), and cell-free culture supernatant (CFS). The LBC was prepared from bacterial suspensions that were centrifuged at 10,000 × g for 10 min at 4 °C, washed three times with sterile PBS, and adjusted to a live bacterial concentration of 107 CFU/mL. The HKC was generated by subjecting L. helveticus to heat treatment at 121 °C for 15 min after the cells were washed three times with sterile PBS and adjusted to a cell density of 107/mL. The CFS was obtained by centrifugation of a comparable amount (~ 107 CFU/mL) of L. helveticus cultivated in MRS broth, and then passing the supernatant through a sterile filter with a pore size of 0.22 μm.
A mucosa-associated E. coli strain belonging to phylogroup B2, characterized by its adhesive and invasive properties, was isolated from the ileal biopsy of a patient with active CD in our previous research [19], was propagated in Luria-Bertani (LB) broth (Merck, Germany).
Cell culture conditions
Caco-2 cell line (ATCC® HTB-37TM), purchased from the Iranian Biological Resource Center, Tehran, Iran, was cultured in 25 cm2 culture flasks until they reached confluent growth at a temperature of 37 °C. The culture medium used was Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Life Technologies, Carlsbad, CA, USA), supplemented with 10% inactivated fetal bovine serum (FBS) (Euroclone, Milan, Italy), 1% nonessential amino acid (NEAA), 1% L-glutamine, and 1% penicillin-streptomycin (Gibco Life Technologies, Paisley, UK). In order to maintain optimal conditions, all cultures were incubated at 37 °C in a humidified atmosphere consisting of 5% carbon dioxide (CO2) and 95% air. When the cells achieved 80–90% confluence, they were dissociated for further passaging using a solution consisting of 0.05% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) at a ratio of 1:3.
Cytotoxicity assay of HA-A. Platensis
The assessment of cytotoxicity of HA-A. platensis on Caco-2 cells was carried out through the MTT colorimetric assay. Initially, Caco-2 cells were seeded in a 96-well plate at a density of 62.5 × 103 cells per well and were allowed to incubate overnight. The cells were then treated with extracts dissolved in a fresh medium for 24 h. HA-A. platensis concentrations (0.125–8 mg/mL) were selected based on published efficacious ranges for intestinal cells [20, 21] and confirmed via pilot viability assays. Subsequently, the cells were exposed to MTT solution at 0.5 mg/mL for 4 h. To solubilize formazan crystals produced by viable cells, the culture medium was replaced with 100 µL of dimethyl sulfoxide (DMSO). Absorbance readings were taken at 570 nm with a reference wavelength of 630 nm. Cell viability was calculated using the formula described previously [22]. For the negative control, cells were solely incubated in a culture medium.
Co-culture of CD-associated E. coli, L. helveticus, and HA-A. Platensis
To assess the potential anti-inflammatory effects of HA-A. platensis and L. helveticus on inflamed colonic cells induced with CD-associated E. coli, Caco-2 cells were cultured in 24-well plates at a concentration of 3 × 106 cells per well and allowed to attach for at least 48 h before the addition of bacteria. Following the incubation period, cells were washed twice with PBS, and then fresh DMEM medium, HA-A. platensis in the concentration of 2 mg/mL or/and L. helveticus at the multiplicity of infection (MOI) 50 (bacteria: cells) were added and incubated at 37 °C in 5% CO2 for two hours. Subsequently, CD-associated E. coli was added at MOI 10 (bacteria: cells), and incubated at 37 °C in 5% CO2 for a further four hours. The cells were washed three times with PBS and then fresh DMEM medium was replaced followed by 48 h incubation at 37 °C in 5% CO2. Moreover, cells treated with sterile MRS medium and untreated cells were designated as the negative control groups. Each trial was conducted in triplicate to guarantee technical reproducibility.
Effect of L. helveticus/HA-A. Platensis on CD-associated E. coli adhesion and invasion
Following treatment, a portion of the cells underwent a triple wash with PBS buffer to eliminate non-adherent bacteria, followed by trypsinization using trypsin–EDTA (0.25% trypsin, 1 mM EDTA). The resulting cell suspension was then subjected to serial tenfold dilutions and spread-plated on LB agar, followed by overnight incubation at 37 °C to enumerate bacterial colonies, specifically focusing on CD-associated E. coli colonies. The anti-adhesion properties of L. helveticus and/or HA-A. platensis were examined through the determination of the CD-associated E. coli adhesion rate (%). This rate was calculated as one minus the ratio of the number of adherent bacteria co-cultured with L. helveticus/HA-A. platensis to the number of adherent bacteria in the control group, multiplied by 100.
A subset of the treated cells was subjected to a triple wash with PBS buffer to eliminate non-adherent bacteria, followed by the addition of 1 mL of gentamicin (200 µg/mL) to eradicate non-invasive E. coli cells. Following this, culture plates were placed in a CO2 incubator at 37 °C for an additional hour to eradicate adhered non-invasive E. coli cells from the Caco-2 cells. Subsequently, the mixed culture medium was aspirated, and the cells were rinsed twice with PBS buffer, then 1 mL of 1% Triton-X 100 was introduced for a 10-minute incubation period to lyse the Caco-2 cells. Serial dilutions of the lysed Caco-2 cells were spread-plated on LB agar and incubated overnight at 37 °C to enumerate bacterial colonies. The anti-invasion properties of L. helveticus and/or HA-A. platensis were investigated by assessing the CD-associated E. coli invasion rate (%). This rate was derived by subtracting the ratio of invasive bacteria co-cultured with L. helveticus/HA-A. platensis from one, and then multiplying the result by 100.
RNA extraction and quantitative real-time PCR (qPCR) analysis
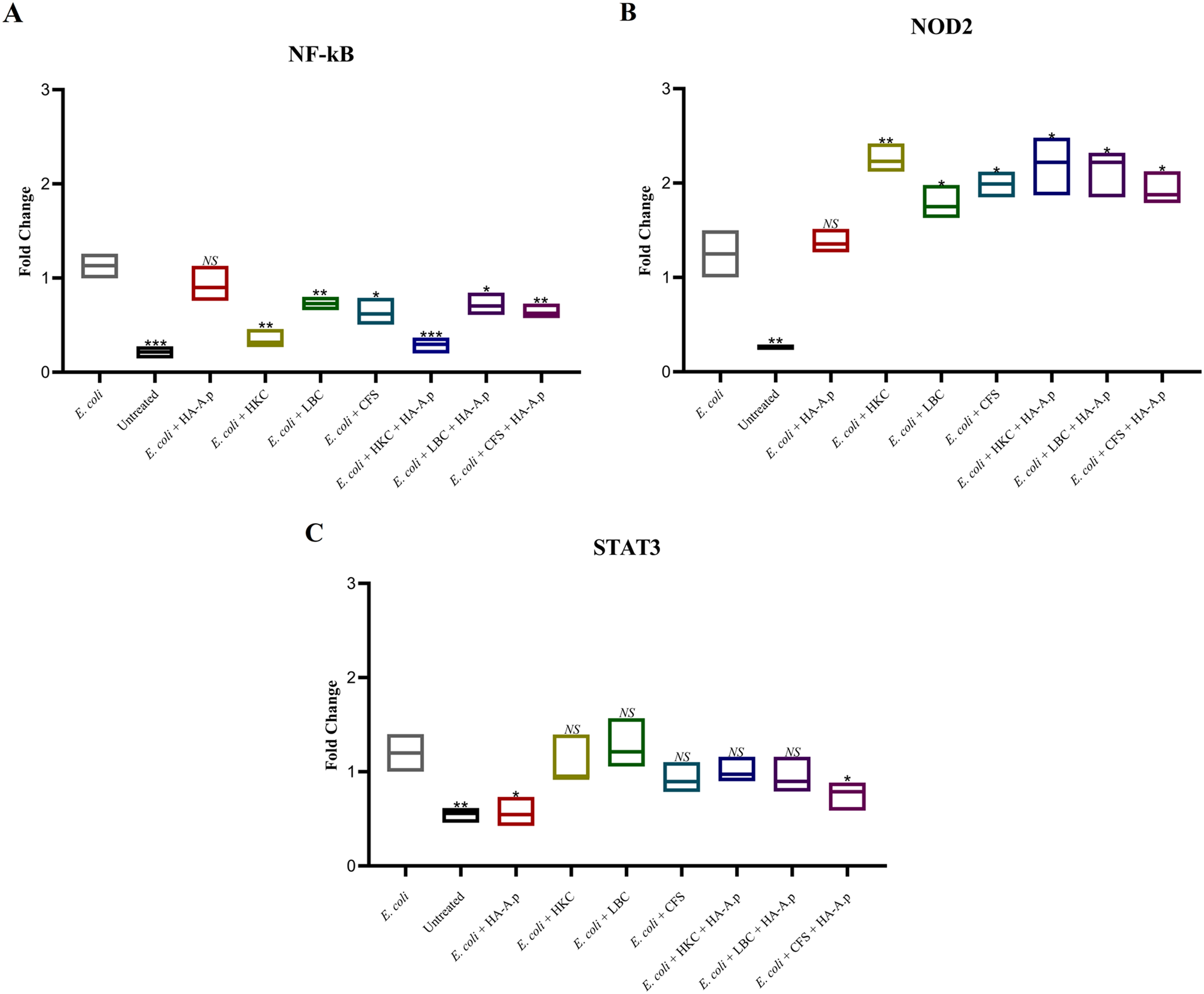
For assessing the expression levels of NF-kB, STAT3, and NOD2, total RNA extraction from cultured cells was executed utilizing the Total RNA Purification Mini kit (Yekta Tajhiz Azma, Iran) as per the prescribed protocol. The quality of the extracted RNA was evaluated through nanodrop spectrophotometry (by analyzing the 260/280 nm and 260/230 nm ratios). Subsequent to this, reverse transcription (RT-PCR) was implemented employing the 2-step 2X RT-PCR Premix (Taq) kit (BioFact™, South Korea) following the manufacturer’s guidelines. Quantitative real-time polymerase chain reaction (qPCR) was conducted in a total volume of 20 µL utilizing the SYBR Green Master Mix (BioFact™, South Korea) on the LightCycler® 96 System (Roche Applied Science, Germany). The qPCR cycling parameters included an initial denaturation at 95 °C for 600 s, followed by 40 cycles of two steps: denaturation at 95 °C for 15 s and amplification at 60 °C for 60 s, along with a single fluorescence measurement. For normalizing the expression levels, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was applied as a housekeeping reference gene in every qPCR run. The primer sequences used in the investigation are disclosed in Table 1. The relative mRNA levels were computed utilizing the 2−ΔΔCt method (where ΔCt = Ct target – Ct housekeeping). All samples were analyzed in triplicate to ensure statistical reliability.
Table 1 Oligonucleotide primers used in this studyQuantification of cytokine concentrations
Upon stimulation, cell culture supernatants (conditioned media) were collected to determine the levels of TNF-α, IL-1β, IL-8, and IL-10, enzyme-linked immunosorbent assay (ELISA) colorimetric kits (Carmania Parsgen Co, Iran) were employed, following the guidelines provided by the manufacturer.
Statistics
All experiments were performed in triplicate (n = 3 biological replicates). Data are presented as mean ± standard deviation (SD). Normality was confirmed using Shapiro-Wilk tests. For comparisons between multiple groups, one-way or two-way ANOVA (for combination treatments) with Tukey’s post-hoc test was applied, as appropriate. Student’s t-test was used for pairwise comparisons when justified. Synergistic effects were analyzed via two-way ANOVA with interaction terms, comparing observed combination effects to expected additive effects. For qPCR data, the 2−ΔΔCt method was used with GAPDH normalization. All statistical tests were two-tailed, with P < 0.05 considered significant. Analyses were performed using SPSS v22 (IBM) and GraphPad Prism v6.07.






Comments (0)