Clinical sample collection
Twelve pairs of BC tissues and paracancerous tissues were collected from the hospital and immediately stored in − 80°C liquid nitrogen. None of the patients received chemotherapy or radiotherapy before tumor resection. Informed consent was obtained from all individual participants prior to any study-related procedure. The characteristics of BC patients are shown in Table 1..
Table 1. Characteristics of patients with BC
Cell culture
The cell lines used in this experiment, normal human bladder epithelial cells (SV-HUC-1 cells) and bladder cancer cells (RT-4 cells, KK47 cells, T24 cells, 5637 cells, HT-1376 cells), were all purchased from Wuhan Pricella Biotechnology Co., Ltd. Wuhan, China. The cells were free of mycoplasma infection, as identified by STR. All cells were cultured in McCoy’s 5 A (modified) medium (Gibco, Invitrogen, Waltham, CA) supplemented with 10% fetal calf serum, 1% penicillin, and streptomycin in an incubator at 37°C and 5% CO2. Routine culture and subculture were performed when the cell density reached 90%. To detect the effect of the AKT signaling pathway, we added 10 μM SC79 (AKT activator) to the T24 cells in the groups.
Animals and experiments
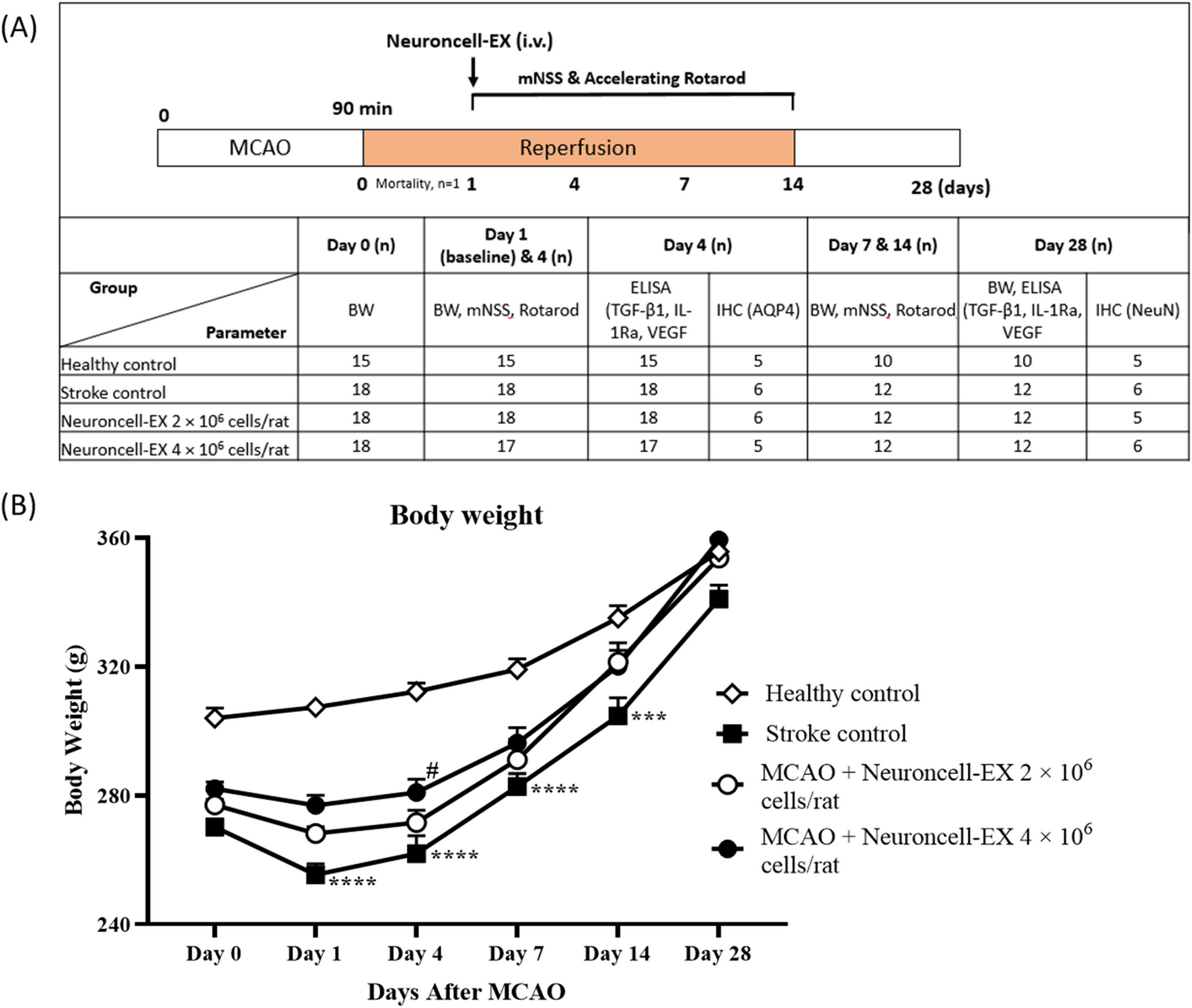
Thirty BALB/c nude mice (weight, 14–16 g) aged 4–6 wk were obtained from the Animal Experiment Center of Kunming Medical University. After the experimental animals were adapted for 1 wk, they were randomly divided into 3 groups (n = 10 each). Normal or T24 cells transfected with miR-944 mimic or oe-SHMT1 were subcutaneously injected into BALB/c nude mice at a cell number of 1 × 107 per nude mouse. From day 5, tumor size was measured every 3 d, and the tumor volume was calculated as (length × width2)/2. After 20 d, the animals were euthanized, and the tumor tissues were collected for the measurement of relevant indicators.
Lentiviral transfection
Lentiviral vectors were purchased from Hunan Fenghui Biotechnology Co., Ltd. Changsha, China for lentiviral transfection. Eighteen to twenty-four h before transfection, adherent cells were spread into 24-well plates at 1 × 105/well. Polybrene was added to the level of 6 μg/mL, and an appropriate amount of virus was added to the suspended cells at 2 × 105/mL, mixed evenly, and incubated at 37°C for 4 h. Then, 2 mL of fresh medium was added to dilute the polybrene. Culture was continued for 3–4 d, and then the cells were passaged or the medium was changed, depending on the cell growth status.
Dual-luciferase experiment
The targeted binding sites of miR-944 and SHMT1 were predicted using the bioinformatics website https://starbase.sysu.edu.cn/. The SHMT1 3′-UTR containing the miR-944 binding site was cloned and inserted into the pGL3 vector (Promega, Madison, WI) to construct the wild-type SHMT1 vector (WT). Mutant SHMT1 vectors (MUT) were generated using a site-directed mutagenesis kit (Stratagene, La Jolla, CA). WT or MUT and overexpression of miR-944 or a negative control were transfected into 293 T cells using Lipofectamine 3000. After 48 h, luciferase activity was detected using a dual-luciferase reporter assay system (Promega).
Real-time quantitative PCR (RT-qPCR)
Total RNA from normal human bladder epithelial cells, bladder cancer cells, and clinical samples was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA was reverse-transcribed into cDNA using the First-Strand cDNA synthesis kit (Genenode, Wuhan, China), followed by real-time fluorescence quantification using SYBR Green. Real-time fluorescence quantitative PCR was performed using a PCR kit (Solarbio, Beijing, China) using U6 as an internal reference, and results were calculated using the 2−ΔΔCt method (Livak and Schmittgen 2001). Details of the primer sequences are shown in Table 2..
Table 2. RT-qPCR primer sequences
Western blot
Total proteins from normal human bladder epithelial cells, BC cells, clinical specimens, and cancer tissues of nude mice were extracted, separated by 10% SDS-PAGE, and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA). After the membrane was blocked with 5% skim milk for 2 h at room temperature, diluted primary antibody was added and incubated at 4°C overnight. Then, it was conjugated with HRP secondary antibody (1:2000, cat. no. ab205718, Abcam, Cambridge, UK) at room temperature for 1 h. Anti-GAPDH antibody (1:1000, cat. no. ab181602, Abcam) was used as a control. Cell exposure and observation were performed using ECL chemiluminescence solution (Biosciences, Cincinnati, OH). Protein band analysis was performed using ImageJ. The primary antibody information is as follows: anti-SHMT1 (1:1000, cat. no. ab186130, Abcam), anti-E-cadherin (1:10,000, cat. no. ab40772, Abcam), anti-vimentin (1: 5000, cat. no. ab92547, Abcam), anti-fibronectin (1:1000, cat. no. ab2413, Abcam), anti-N-cadherin (1:1000, cat. no. ab76011, Abcam), anti-ATIC (1:2000, A304-272 A, Thermo Fisher Scientific, Waltham, MA), anti-AKT (1:25,000, cat. no. ab81283, Abcam), anti-FOXO3 A (1:1000, cat. no. ab109629, Abcam), anti-Beclin1 (1:2000, cat. no. ab207612, Abcam), anti-p62 (1:1000, cat. no. ab109012, Abcam), anti-LC3B (1:1000, cat. no. ab192890, Abcam).
Detection of cell proliferation by CCK-8
Detection was performed using a CCK-8 detection kit (Beyotime, Shanghai, China). T24 cells were seeded in 96-well plates at a concentration of 5 × 103 cells/well. They were grouped according to the experimental requirements, and three replicate wells were set up for each group. After performing the experiment according to the requirements of each group, the original culture medium was discarded, and after the addition of new 100 μL cell culture medium, 10 μL CCK-8 solution (Beyotime, Beijing, China) was added and incubated at 37°C in the dark for 2 h. All absorbance values were measured at 450 nm using a microplate reader.
Scratch experiment
T24 cells in each group in the logarithmic growth phase were collected. After the cells were normally digested and passaged, the cells were seeded on 24-well plates. When the cell density reached 90%, they were scratched with a pipette tip. After the cells were cultured for 24 h, cell migration was observed under a microscope and photographed.
Transwell experiment
Cell invasion experiments were performed in Transwell wells (Corning, Corning, NY). T24 cells in each group were added to serum-free McCoy’s 5 A medium to adjust the concentration to 1 × 105 cells/mL. Two hundred microliters of cell suspension was added to the upper chamber of a Matrigel-coated Transwell. In the lower chamber of the 24-well plate, 600 μL of McCoy’s 5 A medium containing 10% fetal calf serum was added. After 24 h of culture, the cells in the lower chamber were fixed with 4% paraformaldehyde, stained with crystal violet (Solarbio, Beijing, China), and observed under an inverted microscope (Olympus, Tokyo, Japan) The number of cells at fixed positions in each well was observed, and five fields were selected to count and take pictures.
Colony formation experiment
T24 cells were seeded in 6-well plates at a density of 1 × 103 cells/well and then cultured continuously for 2 wk to form colonies. Five milliliters of 4% paraformaldehyde was added to each well and incubated for 15 min. Subsequently, colonies were fixed with methanol and stained for 15 min with a Giemsa staining kit (Solarbio, Beijing, China). After washing with tap water, colony counting was performed (Du et al. 2018), and the experiment was repeated three times.
Coimmunoprecipitation
T24 cells were harvested and lysed in IP lysis buffer, and the supernatant was collected by centrifugation. Protein A/G Sepharose (Santa Cruz Biotechnology, Santa Cruz, CA) was incubated with anti-SHMT1 antibody (1:200, cat. no. ab186130, Abcam) and anti-ATIC antibody (1:200, A304-272 A, Thermo Fisher Scientific, Waltham, MA). All IPs were preincubated at 4°C for 60 min with gentle shaking; all IPs were incubated at 4°C overnight with slow shaking; and the beads were collected by centrifugation and washed three times with lysis buffer. The immunoprecipitates were subjected to western blot analysis.
Monodansylcadaverine (MDC) Labeling
A cell autophagy staining detection kit (MDC method) was purchased from Shanghai Beyotime (cat. no. C3018S). After transfected T24 cells from each group were seeded in 24-well culture plates at 3 × 104 cells per well, 250 µL of MDC (1 ×) was added to the cells for 48 h. Then, the cells were incubated at 37°C and 5% CO2 in the dark for 30 min. The MDC staining solution was aspirated off, the cells were washed three times with assay buffer, and then 1 mL of assay buffer was added. The samples were excited with UV excitation light under a fluorescence microscope, and green fluorescence was observed. Alternatively, fluorescence detection was performed using a fluorescence microplate reader at a wavelength of 335 nm.
Immunohistochemical staining for ki-67
Paraffin-embedded bladder cancer tumor bodies were deparaffinized and rehydrated, and then antigen retrieval was performed in 0.01 M citrate buffer (pH 6.0). Slides were incubated with ki-67 (1:100, cat. no. ab15580, Abcam) at 4°C overnight, and immunodetection was performed using HRP secondary antibody and DAB chromogenic reagent. The results were photographed under a microscope (NiKon, Tokyo, Japan), and quantification of IHC staining density was measured using ImageJ software.
Detection of apoptosis levels
Flow cytometry was used to determine the level of surface apoptosis, and T24 cells were first harvested, digested with trypsin, and washed with PBS. Cells were stained according to the operating instructions of the Annexin V-FITC/PI Apoptosis Detection kit (Invitrogen, Carlsbad, CA) and incubated at 25°C in the dark for 15 min. After the completion of incubation, the level of apoptosis was measured by flow cytometry (FACScan, Temecula, CA).
Statistical analysis
All experimental data in this paper are expressed as the mean ± standard deviation (mean ± SD). The data were analyzed and graphed using GraphPad Prism 7. t tests were used for comparison between two groups, and one- or two-way analysis of variance (ANOVA) was used for multigroup comparisons, followed by Tukey’s HSD test. P < 0.05 indicated that the difference was statistically significant.




Comments (0)