
Remember me





The immune system is typically divided into two complementary parts: innate immunity and adaptive immunity. These two branches differ significantly in the types of immune cells involved, the speed of activation, the specificity of responses, and the capacity for immunological memory. Innate immunity rapidly recognizes and responds to pathogens or damage-associated molecular patterns (PAMPs or DAMPs) through pattern recognition receptors (PRRs), thereby providing immediate and effective host defense [1]. However, these responses are relatively non-specific, and traditional views suggest that they do not generate immune memory. In contrast, adaptive immunity develops more slowly, often taking days to weeks to fully engage, triggered by the recognition of specific antigens, ensuring a high degree of specificity against pathogens. The activation of the adaptive immune system is facilitated by specialized antigen-presenting cells from the innate immune system, leading to the formation of immunological memory that guarantees long-term protection [2]. Immune memory has been considered exclusive to the adaptive immune system in the past. More recently the concept of trained immunity has gained traction, demonstrating that the innate immune system can also establish long-lasting immune memory following infections and certain vaccinations, thus enhancing responsiveness upon re-encountering with the same or even unrelated pathogens [3]. This concept is referred to as “trained immunity”.
In contrast with adaptive immune memory which is based on specific antigens and relies on genetic rearrangement mechanisms as well as clonal expansion of adaptive immune cells, trained immunity is fundamentally linked to changes in cellular reprogramming of innate immune cells [4]. These alterations are intricately associated with metabolic reprogramming and epigenetic modifications that regulate the transcription of genes related to host defense upon reactivation. The functional reprogramming of innate immune cells leads to more effective antimicrobial responses, such as enhanced phagocytosis and cytokine secretion, increased production of reactive oxygen species (ROS), and more efficient pathogen clearance mechanisms [5]. Given the short lifespan and non-proliferative nature of innate immune cells, researchers have proposed that persistent innate immune memory may be mediated by state and fate adaptation in long-lived immune stem cells (hematopoietic stem cells, HSCs) [6]. HSC-mediated durable innate immune memory is termed “central trained immunity” [7].
HSCs are the common progenitor cells for all immune cells, which finely balance self-renewal state and multi-lineage differentiation fate to adapt to immune demands. Previous study demonstrated that HSCs directly participate in the primary response to both acute and chronic infections through increasing proliferation to replace depleted progeny pools [8,9,10]. In addition to the passive “pushed” effect that peripheral cell deficiencies may exert on HSCs, HSCs can also be active “pull” towards cell division and differentiation fate by directly sensing infection [11]. Typically, innate immune cells serve as the first line of defense in the organism, requiring widespread distribution or constant patrol within various peripheral tissues to recognize invading pathogens. HSCs residing in the bone marrow were previously considered to exhibit a delayed response to pathogen detection. However, recent research has revealed that when microbial invasion poses a severe threat to life, HSCs can directly recognize and respond to infections [12]. Infections activate downstream targets and reprogram HSC state and fate through both intrinsic and extrinsic immune signaling pathways, such as Toll-like receptors (TLRs), DNA/RNA sensors, and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), leading to hematopoietic-immune stress and the release of robust immune alert signals [13]. Concurrently, pathogen-derived compounds can trigger stress-induced hematopoiesis, characterized by HSC proliferation and enhanced myeloid differentiation [14]. Therefore, perturbation of HSC state and fate leads to systemic effects in immune responses.
Recent advances have demonstrated that, under certain circumstances, immune response can induce persistent innate immune memory in hematopoietic stem and progenitor cells (HSPCs), thereby enhancing broad resistance to secondary challenges [15,16,17,18]. These investigations elucidate the activation of cell proliferation and myeloid hematopoiesis pathways following immune challenges driven by microorganisms (such as Bacillus Calmette-Guérin, BCG and Mycobacterium tuberculosis, Mtb), microbial compounds (such as Lipopolysaccharide, LPS and β-glucans), viral mimetics (such as polyinosinic: polycytidylic acid, Poly: IC) and other factors (such as heme and Western diet) [19]. In these scenarios, the initial exposure to stimuli triggers long-term epigenetic and metabolic reprogramming of HSPCs. HSC state and fate in trained immunity are characterized by an increased quantity of myeloid cell production, enhanced non-specific antibacterial capabilities in HSC progeny cells, and heightened stress resistance of HSCs to secondary infections or chemotherapy-induced bone marrow suppression [16, 17]. Importantly, these phenomena have also been observed in human HSCs (hHSCs), with evident immune memory characteristics in HSCs and progeny cells following BCG vaccination [20, 21]. Otherwise, previously unrecognized inducer of trained immunity like labile heme could also confer long-term immunological regulation on hHSCs [22]. Western diet has been found to reprogram HSPCs, resulting in deleterious trained immunity and increasing the reactivity of innate immune cells [23]. Thus, HSCs de facto serve as the central hub of trained immunity, while the effects and mechanisms through which HSCs participate in the trained immune response, along with the alterations in their state and fate in both adaptive and maladaptive trained immunity, remain not fully understood.
In this review, we summarized the recent advances of HSC state and fate under homeostatic conditions, during acute and chronic infections, and in response to immune modulators, representing key areas of current trained immunity research. This review aims to deepen the understanding of the effects and mechanisms by which stem cell-level regulation influences trained immunity. Additionally, it may offer new strategies for the treatment of infectious and inflammatory diseases, as well as provide fresh perspectives for developing novel immune therapies.
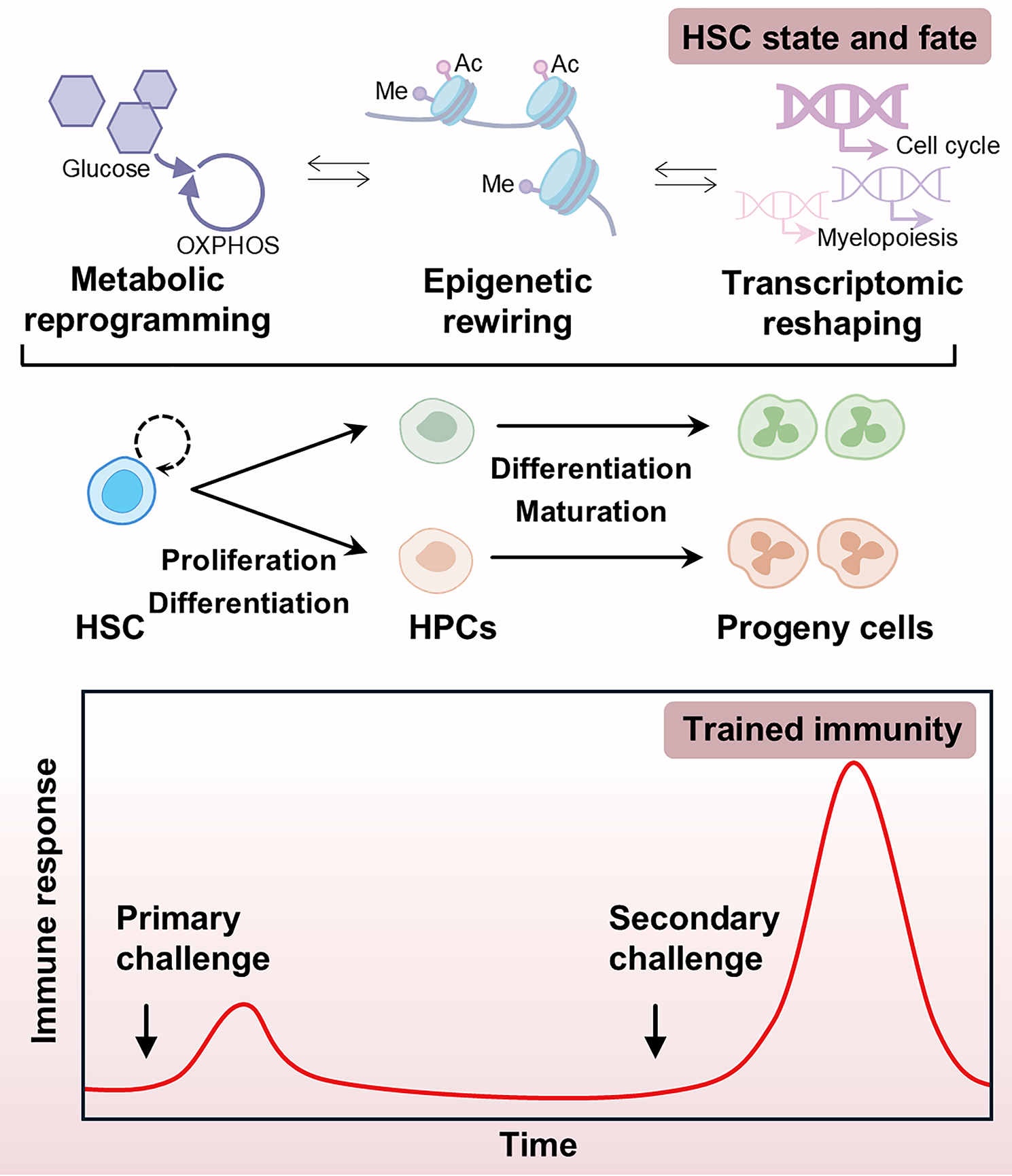
Of note, in the context of trained immunity, changes in HSC “state” predominantly occur at the transcriptional, epigenetic, and metabolic levels, while alterations in HSC “fate” are reflected in self-renewal capacity, proliferative activity, differentiation potential and survival advantage. For clarity, this review adopts the following terminology: (1) HSC state encompasses molecular-level modifications (transcriptional, epigenetic, and metabolic); (2) HSC fate refers to as functional changes in stem cell behavior (self-renewal, proliferation, differentiation and survival). Since current investigations into trained immunity predominantly rely on murine models, this review referenced herein employ these experimental systems unless explicitly stated.
HSC state and fate support immune functionality under homeostasisTo manage the state and fate under homeostasis, HSCs engage a delicate balance of pro-survival and stress-response mechanisms, ensuring a functional HSC compartment while adapting to the demands of immune cell production. In response to stress, HSCs activate survival mechanisms to safeguard HSC populations. For instance, HSCs display greater anti-apoptotic capacity compared to downstream progenitor cells under stress, evidenced by reduced pro-apoptotic gene expression and increased pro-survival gene expression [24], quiescence-mediated protection from the p53-dependent apoptotic program [25], and by survival advantages conferred by microenvironmental cells [26]. In addition, HSCs maintain self-renewal and regenerative potential via various stress-response pathways, including autophagy and mitophagy. Autophagy helps preserve HSC quiescence by degrading active mitochondria and inhibiting differentiation signals from oxidative phosphorylation (OXPHOS), thereby enabling a shift back to glycolytic metabolism upon cell cycle activation [27, 28]. Mitophagy, executed through peroxisome proliferator-activated receptors, further supports quiescence by removing damaged mitochondria [29]. HSC fitness also depends on tightly regulated protein synthesis rates, which protect HSCs from functional deficits induced by the accumulation of misfolded proteins [30]. By utilizing intrinsic mechanisms, HSCs maintain quiescence and protect against the effects of active metabolism in the hypoxic bone marrow. Quiescent HSCs primarily rely on glycolysis for energy, minimizing mitochondrial respiration and ROS production associated with OXPHOS [31]. Of note, fine-tuned metabolic activation of HSCs also undergoes epigenetic and transcriptional remodeling, which are crucial for HSC self-renewal and fate decision [32]. Thus, the enforcement of relatively quiescent state and stable fate in HSCs is a crucial characteristic, which protects the HSC pool integrity and lifelong immune cell production.
HSCs keep their state and fate during steady-state hematopoiesis, but play a crucial role in responding to stresses such as hemorrhage, infections, and chemotherapy or radiotherapy. The antiviral cytokines Type I interferon (IFNα) drives HSC proliferation by briefly relaxing quiescence-enforcing mechanisms in response to acute IFNα exposure [25]. Nevertheless, this proliferative response is transient, with HSCs quickly returning to a protective quiescent state. It has been demonstrated that IFNα could prime HSCs for apoptosis but induce direct cell death only upon active proliferation, thereby explaining their suppressive effects on HSC function [25]. The inflammatory cytokine tumor necrosis factor-alpha (TNF-α) shield HSCs from depletion during inflammation by activating the transcription factor PU.1, which inhibits excessive cell cycle progression and protein synthesis [33]. Overall, HSCs dynamically respond to inflammatory stress while maintaining their numbers and functions, thus preserving homeostasis in the hematopoietic-immune system throughout the lifespan. Despite such HSC responses can be beneficial in promoting the elimination of an acute infection, chronically sustained activation may impair HSC function, ultimately leading to HSC exhaustion and persistence of inflammatory pathologies. For instance, prolonged or repeated infections may disrupt this quiescent state, causing excessive proliferation or abnormal differentiation of HSCs, which can ultimately lead to premature exhaustion or depletion of the HSC pool [34]. Therefore, the state and fate of HSCs direct the production and function of their progeny immune cells, which in turn determines the overall immune functionality of the organism.
HSC state and fate in beneficial and maladaptive trained immunityTrained immunity reshapes the transcriptomic, epigenetic, and metabolic states of HSCsHSCs undergo continuous adaptations in transcription, epigenetics, and metabolism, which provides a foundation for the induction of trained immunity. Similar to observations in monocytes and macrophages, the establishment of immune memory in HSCs is closely linked to alterations in their transcriptomic landscape. Analysis following initial stimulation with β-glucan and BCG revealed evident activation of gene programs associated with the cell cycle and myeloid differentiation within HSC lineages [15,16,17]. Specifically, BCG reprograms HSCs through IFN-γ signaling, promoting their priming and expansion [15]. β-glucan induces HSC training through the IL-1β/GM-CSF axis with adaptations in glucose metabolism and cholesterol biosynthesis [16]. Trained immunity induced by Listeria monocytogenes is manifested by significant increased HSCs and multipotent progenitors (MPPs), including increased myeloid-biased MPP3 and lymphoid-biased MPP4, which contributes to the elevated leukocyte counts before and during infection [17]. Following LPS stimulation, HSCs displayed transient changes in their abundance, progeny, composition and gene expression. However, persistent alterations were observed in the C/EBPβ-dependent accessibility of specific myeloid lineage enhancers, which heightened the responsiveness of associated immune genes to secondary stimulation [18]. It demonstrated that LPS-induced gene transcriptional changes, rather than epigenetic alterations, are not sustained, as HSCs quickly revert to transcriptional homeostasis [18]. In this context, epigenetic alterations facilitate the persistent presence of previously dormant open chromatin regions in innate immune genes, which are primed for reactivation upon secondary stimulation.
Trained immunity is influenced by a combination of metabolic and epigenetic adaptations, which are often functionally interconnected (Fig. 1). Following BCG vaccination, macrophages derived from BCG-trained HSCs exhibit accessible chromatin at interferon regulatory factor (IRF) and signal transducer and activator of transcription (STAT) binding sites, thereby establishing a long-lasting antimicrobial immune memory. These changes can persist for several weeks in the absence of BCG stimulation and are associated with stable modifications in activating histone marks, including mono- and trimethylation of H3K4 and acetylation of H3K27 [15]. In the β-glucan stimulation model, specific changes in gene programs regulating cellular metabolism were observed in HSCs. It was indicated that innate immune memory in HSCs is associated with a shift toward glycolytic metabolism and cholesterol biosynthesis, particularly through the mevalonate pathway, thereby mimicking key metabolic features related to monocyte/macrophage immune training [16]. In contrast, dysregulation of iron metabolism in HSCs following Mtb infection hinders myeloid differentiation and directly drives the development of a tolerant phenotype [35]. Furthermore, cholesterol accumulation has been shown to promote myelopoiesis as a result of trained immunity in HSCs [16]. Intriguingly, BCG vaccination persistently alters gene expression in hHSCs. In contrast, it modulates chromatin accessibility in hematopoietic progenitor cells (HPCs), with the most prominent changes occurring at sites regulated by Kruppel-like factors (KLF) and early growth response (EGR) transcription factors, rather than affecting gene expression [21]. In this way, HSCs maintain a differentiation bias toward the myeloid lineage after 3 months post BCG vaccination [21]. This finding suggests that chromatin accessibility serves as an epigenetic memory in HSCs in trained immunity, which is subsequently imparted onto the progeny cells, even if many of the differential gene expression programs are silenced. Overall, adaptations in transcription, epigenetics, and metabolism in HSCs lay the groundwork for trained immunity induction.
Fig. 1
Trained immunity reshapes the state and fate of HSCs. Trained immunity agonists stimulate long-term metabolic, epigenetic, and consequent transcriptional adaptations in HSC state and fate. The trained HSCs proliferate and preferentially give rise to trained progeny immune cells. The trained phenotype enables innate immune cells to respond faster and stronger to secondary challenges with the same or heterologous stimuli
Trained immunity profoundly impacts the fate of HSCs and their progeny cellsPersistent fate remodeling of HSPCs induced by trained immunity preferentially occurring in more primitive HSCs has been observed in both mouse models and human samples, which is characterized by HSC proliferation and myeloid differentiation (Table 1; Fig. 2) [15, 21]. This preference may be explained by heterogeneity of HSCs, as trained immunity predominantly induces selective expansion of myeloid-biased HSCs, leading to their clonal advantage [16]. On the contrary, Mtb infection exclusively induced cell death in myeloid lineage HSPCs, which impaired trained immunity against subsequent Mtb infection [35]. Trained immunity also educates a subset of HSCs that have always existed but were recently discovered. For instance, Mycobacterium avium (M. avium) induced trained immunity in HSCs and directed the expansion of a distinct HSC subpopulation with an infection-activated phenotype [36], a concept that might prove true across species [37]. This HSC subpopulation expressed not only classical HSC markers but also upregulated markers associated with activated immune responses to infection and those related to B cells [36]. Future studies, incorporating advanced single-cell tracing and clonal analysis techniques, may determine whether the long-term impact of trained immunity is initiated at the level of cell fate decision in primitive HSCs and lineage-committed subpopulations.
Table 1 HSC state and fate in trained immunityFig. 2
Overview of HSC state and fate in beneficial and maladaptive trained immunity. Current studies suggest that trained immunity can have both beneficial and detrimental effects. BCG vaccination and β-glucan administration are known to induce a long-lasting reprograming of myeloid cells, resulting in the unspecific protection against different pathogens. Otherwise, previously unrecognized inducer of trained immunity like labile heme could also confer long-term immunological regulation on HSCs. On the other side, western diet was found to reprogram bone marrow granulocyte progenitors, increasing the inflammatory reactivity of innate immune cells. Similarly, maladaptive trained immunity was suggested to be involved in the pathological hyperinflammation affecting patients suffering from CHIP and cardiovascular diseases. A healthy trained immunity response that is essential for immune homeostasis provides the unspecific protection against pathogens. However, maladaptive trained immunity can lead to a variety of inflammatory and autoimmune diseases
One of the outstanding questions in trained immunity is the longevity of imprinting in trained immune cells. The concept of trained immunity being initiated at the stem cell-level is further supported by analyses of HSC responses in healthy volunteers vaccinated with BCG [20]. BCG vaccination induced a sustained myeloid bias in the transcriptome of hHSCs, accompanied by a lasting (post 3 months) enhancement in the responsiveness of peripheral innate immune cells to heterologous stimuli [20]. Transplantation experiments confirmed that BCG-exposed HSCs displayed enhanced myeloid hematopoiesis even 12 weeks post transplantation [16]. A more recent study utilizing single-cell RNA sequencing and bone marrow transplantation experiments demonstrated that the imprinting of HSCs by BCG and Mtb contributes to protective or failed trained immunity for at least 1 year, respectively, and can be transmitted to progeny cells [35]. Additionally, competitive bone marrow transplantation experiments indicated that BCG-trained HSCs exhibit superior engraftment compared with control or Mtb-exposed HSCs, indicating that BCG reprograms HSCs to enhance their engraftment capacity [35]. Nevertheless, transplantation of M. avium -trained HSC alone did not result in improved immunity, suggesting that the protective trained immune response is probably dependent on other HSPC-compartment cells [36]. These results suggest that various HSPC components may play distinct roles in mechanisms that could be complementary in trained immunity. Additionally, an intergenerational and transgenerational transmission of C. albicans induced trained immunity in HSPCs was observed across two generations [38]. Strikingly, the progeny of C. albicans-exposed mice inherited an epigenetic signature from the HSPC populations of their parents, resulting in improved myeloid cell output and activation as well as increased survival following Escherichia coli infection [38].
Importantly, trained immunity not only enhances the responsiveness of HSCs themselves to subsequent challenges but also improved the quality and function of their progeny immune cells [6, 11]. Recent advances have further revealed that BCG alters gene expression and chromatin accessibility in hHSCs which predicts cytokine secretion in paired peripheral blood mononuclear cells (PBMCs) [20, 21]. Macrophages derived from M. avium-trained HSPCs exhibited enhanced bacterial killing and metabolism [36]. These studies suggest that the immune memory characteristics and functional properties of HSCs can be conveyed to their progeny immune cells through hierarchical differentiation. However, the specific molecular mechanisms underlying this process remain to be elucidated.
Otherwise, the significance of the observed alterations of HSC fate in various memory-inducing factors in these studies still requires further confirmation. The complex interplay among the type, intensity, and duration of the initial stimulus determines the acquisition of memory and its characteristics, influencing either the enhancement (pre-activation) or suppression (tolerance) of subsequent immune responses. For instance, exposure to low and high doses of LPS can respectively induce pre-activation or tolerance in macrophages during subsequent challenges [39]. In vivo high-dose LPS stimulation exerts dual effects on HSCs, promoting robust secondary innate immune responses and simultaneously limiting certain potentially harmful inflammatory signals [18]. β-Glucan treatment also promotes granulopoiesis of HSCs in a IFNα-dependent manner, leading to the generation of a distinct neutrophil subset endowed with regulatory properties. These specialized neutrophils are essential for establishing disease tolerance and maintaining lung tissue homeostasis during viral infection [40]. Consistent with this notion, the acquisition of memory and its outcomes are linked to the activation of specific cytokine pathways. For example, IL-1, GM-CSF, and type II interferon (IFNγ) are associated with protective phenotypes following β-glucan or BCG stimulation [15, 16, 41, 42]. Conversely, IFNα activity is related to acquired tolerance induced by Mtb infection [35]. The intricate rules connecting the initial inducers, target cells, and cell-fate outcomes following secondary stimulation remain to be clarified.
Maladaptive trained immunity induced dysregulation of HSC state and fateTrained immunity provides broad protection against reinfection and plays a vital role in other disease contexts, such as reinforcing anti-tumor effects [43]. However, maladaptive trained immunity can also promote dysregulated immune responses, exacerbating chronic inflammation and metabolic diseases, including atherosclerosis and primary hyperaldosteronism, where myeloid cells are pivotal to the pathogenesis [11]. Maladaptive trained immunity can be induced by various inflammatory stimuli, which results in a persistent inflammatory state through the increased production of myeloid cells with remarkably proinflammatory potential (Table 1; Fig. 2). These observations were consistent with transcriptomic alteration in HSCs, which demonstrated that the enrichment of innate immune-related pathways at the expense of pathways was related to lymphocyte hematopoiesis and function [
Comments (0)