
Remember me
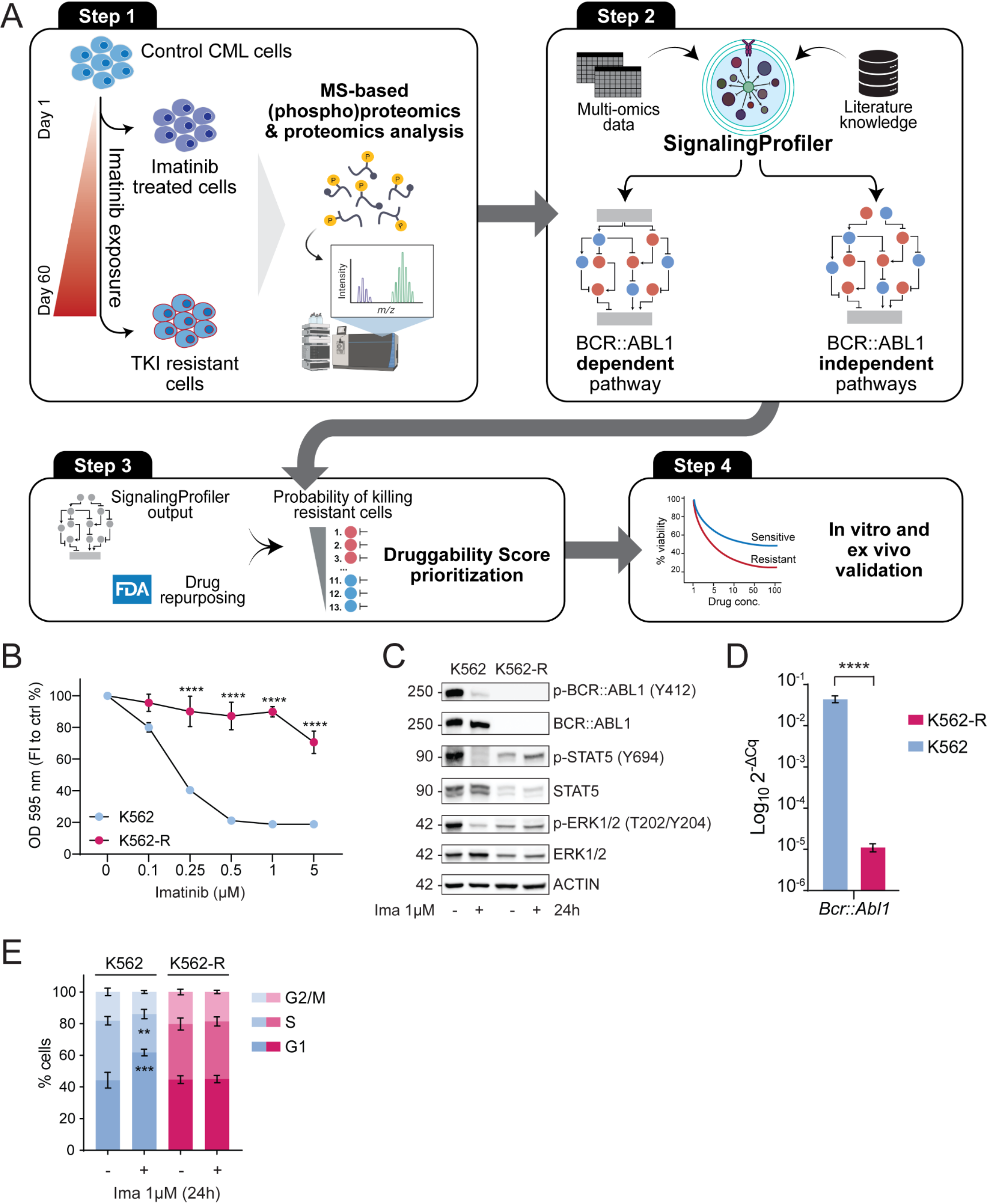





Utilizing the knowledge base INDRA database to assign normalized knowledge scores for all protein kinases, we identified 19 understudied or “dark” protein tyrosine kinases (Supplementary Fig. 1A). Out of these 19 “dark” kinases, we selected five kinases to screen using a multiplex functional kinase activity profiling technology (PamStation12 platform). The kinases we profiled include EPHA6, AATK, INSRR, LTK, and TNK1. The normalized ranks for these kinases range from 0.20 to 0.28, where 0 represents the most understudied kinase and 1 represents the most studied kinase (Supplementary Fig. 1B).
Functional profiling of dark tyrosine protein kinasesScreening purified recombinant “dark” tyrosine protein kinases under four conditions (low, medium, high protein concentrations, and heat inactivation as negative control) revealed a set of clusters of concentration-dependent reporter peptides. Based on concentration–response patterns and a semi-supervised clustering approach, we grouped peptides into four different categories: zero or no affinity, low, medium, and high affinity (Supplementary Table 2). This analysis identified 18 high affinity peptides, 31 medium affinity peptides, and 4 low affinity peptides for EPHA6 (Fig. 2B). This analysis identified 26, 9, 3 and 3 high affinity peptides, 31, 46, 4 and 1 medium affinity peptides, and 6, 10, 8 and 1 low affinity peptides for TNK1, AATK, INSRR and LTK respectively (Supplementary Fig. 2A, 2G, 2M, 2S). Signal intensity of phosphosites with increasing total protein (2.5 ng – 25 ng) of recombinant protein shows concentration specific increases in reporter peptide phosphorylation (Fig. 2C, Supplementary Fig. 2D, 2J, 2P, 2V).
Fig. 2
PamStation12 phospho-tyrosine profiles of purified recombinant dark protein kinases identify novel substrates. A Highlighting the five dark kinases (LTK, TNK1, INSRR, AATK, EPHA6) selected for profiling on a kinome phylogenetic tree. B 2.5 ng, 25 ng, 250 ng of EPHA6 total protein was screened along with a 250 ng EPHA6 heat inactivated (denatured protein) control. Red indicates increased phosphorylation and yellow indicates less phosphorylation at that peptide. A semi-supervised clustering and principal component analysis (PCA) is visualized for PamChip peptides reporting on EPHA6. C Signal intensity plot illustrating increased activity with increased total protein of EPHA6. D Peptide sequence logos for EPHA6 peptides identified on the phosphor-tyrosine (PTK) Pamchip. The color of the single letter amino acid code denotes the chemical features of each amino acid map against relative position. High Affinity: list of peptides that show phosphorylation activity at the lowest protein concentration. Medium Affinity: list of peptides that only start showing phosphorylation activity at the medium protein concentration. Low Affinity: a list of peptides that only show phosphorylation activity at the highest protein concentration. No Affinity: a list of peptides that show no phosphorylation activity across all tested protein concentrations. Ephrin Type-A Receptor 6 (EPHA6)
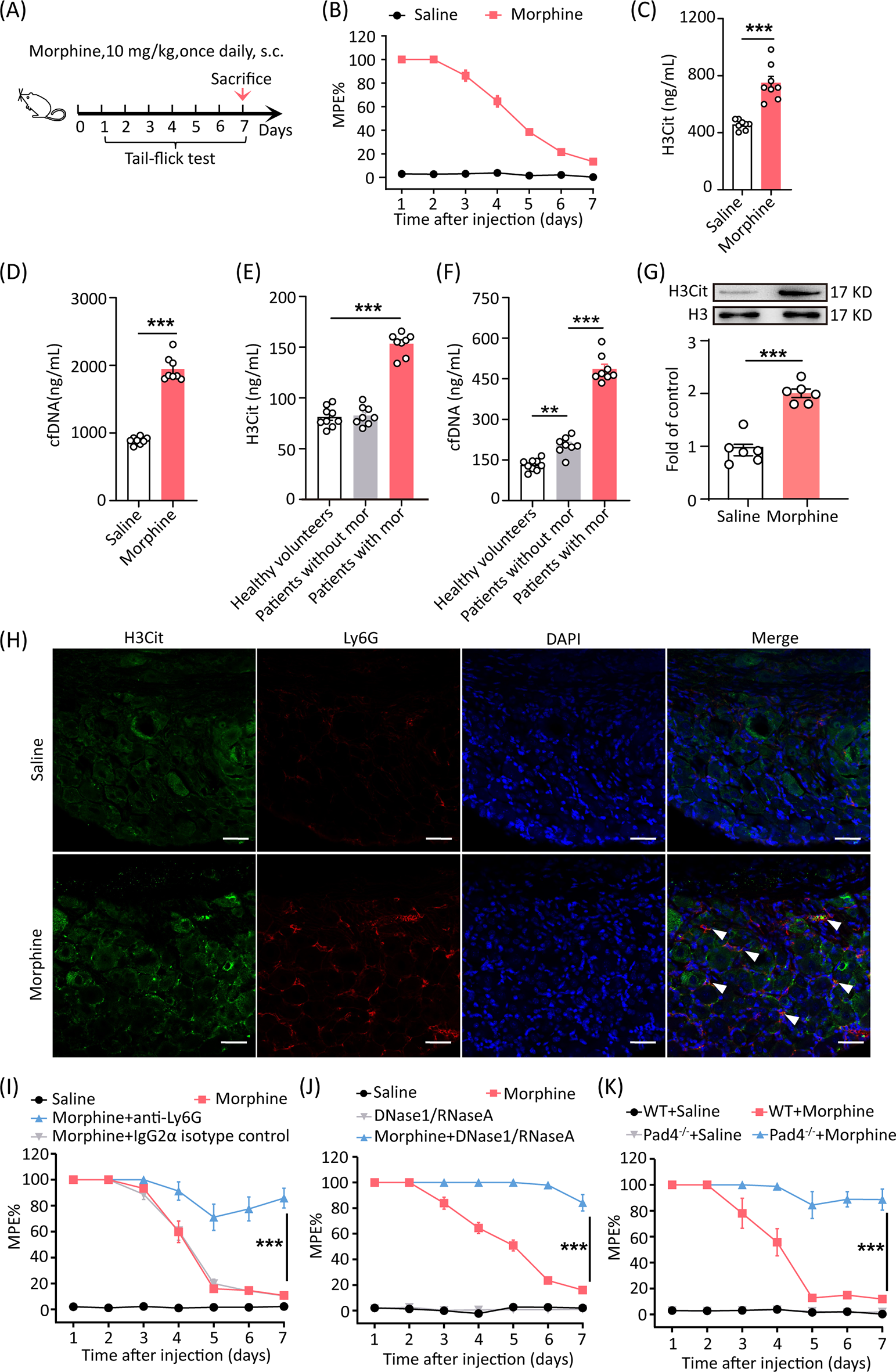
Chip coverage was calculated by dividing the number of peptide hits by the total number of peptides present on the PTK PamChip (193 peptides). We observed 34%, 27%, 8%, 3%, and 33% chip coverage for AATK, EPHA6, INSRR, LTK, and TNK1 kinases, respectively (Fig. 3A). Next, we examined the intersection of peptide hits across all of the kinase profiles and visualized the overlap as an UpSet plot (Fig. 3B) [35]. The chip coverage and peptide overlap analysis reveals the degree of selectivity of our list of kinases to the reporter peptides printed on the PTK PamChip. For example, the relatively low chip coverage for INSRR and LTK suggests that these kinases are more selective than AATK, EPHA6, and TNK1. Additionally, peptide overlap analysis revealed which reporter peptides are uniquely phosphorylated by the recombinant dark protein kinases. For instance, the peptide overlap analysis shows that the majority of the peptide hits of AATK are exclusively phosphorylated by AATK and not by the other kinases. On the other hand, the majority of peptides hits of EPHA6 and TNK1 are shared among the two kinases.
Fig. 3
Chip coverage and peptide overlap analyses reveal differential kinase-substrate selectivity. A Pie charts of chip coverage of AATK, EPHA6, INSRR, LTK, and TNK1. The chip coverage is calculated by dividing the number of peptide hits for each kinase by the total number of peptides present on the protein tyrosine kinase (PTK) PamChip (193 peptides). “Low” denotes low affinity peptides, “Medium” denotes medium affinity peptides, “High” denotes high affinity peptides. B UpSet plot showing the overlap of peptide hits across all recombinant kinase profiles and peptide clusters
Next, we examined the consensus of the amino acid sequence for each cluster of peptides by extracting the main tyrosine phosphosite and the ten neighboring amino acids (five amino acids from each side) of all peptide hits. We visualized the amino acid sequence logos of the three clusters of peptides for each recombinant kinase screening profile (Fig. 2C, Supplementary Fig. 2E, 2K, 2Q, 2W). Pathway analyses were performed using the list of peptide hits for each recombinant kinase screening profile using Enrichr. Using the Gene Ontology (GO) Biological Process 2021 gene set library, we extracted and visualized the top ten pathways (Fig. 2D, Supplementary Fig. 2F, 2L, 2Q, 2X).
In-silico data exploration of dark kinasesKnown and predicted functional pathwaysWe explored several publicly available biological databases to examine the current functional knowledge of our list of kinases in terms of their role as enzymes or substrates, tissue-specific and brain-specific expression at the mRNA and protein levels, known and predicted pathways, subcellular localization, and protein–protein interaction (PPI) networks.
To investigate known downstream substrates for kinases, we queried the iPTMnet database [36]. Unsurprisingly, there are currently no annotated downstream substrates for our list of kinases (Supplementary Table 3). Next, we examined gene expression of our list of targets at the mRNA and protein level using the GTEx and HPA databases across different tissues and specifically in the brain. The results from these databases indicate that AATK, INSRR, and EPHA6 are highly enriched in the brain. Additionally, the HPA database shows INSRR is enriched in the kidney as well. Interestingly, TNK1 shows low brain expression at the mRNA level but high abundance at the protein level. LTK shows overall low expression across all tissues with the highest being in the lung and intestine (Supplementary Table 4).
In terms of subcellular localization, the HPA database also contains immunofluorescent staining images of human cell lines aiming to fully annotate the subcellular localization of the human proteome. Examining the subcellular localization of our selected dark protein kinases reveals differential localization patterns. AATK is localized to mitochondria, EPHA6 to the nucleoplasm, LTK to vesicles such as endosomes and lysosomes, and TNK1 to cell junctions. No data was available for INSRR.
Next, we explored known and predicted functional pathways of our selected dark protein kinases. For known pathways, we utilized Enrichr and the Gene Ontology (GO) Biological Process 2021 gene set library using the HGNC symbol of the kinase as the single input. The results from this analysis showed several brain-specific pathways for some of our kinases. For example, AATK is involved in brain development (GO:0007420) and central nervous system development (GO:0007417). EPHA6 is involved in axon guidance (GO:0007411) and axonogenesis (GO:0007409). LTK is involved in regulation of neuron differentiation (GO:0045664) and regulation of neuron projection development (GO:0010976 and GO:0010975). INSRR and TNK1 have other pathways that include cellular response to pH (GO:0071467) and actin cytoskeleton reorganization (GO:0031532) for INSRR and innate immune response (GO:0045087) for TNK1 (Supplementary Table 5).
Given that the functional knowledge of understudied kinases is relatively low, it is more suitable to examine predicted functional annotations. To investigate predicted functional pathways, we used three approaches: co-expression clustering, protein–protein interaction networks, and genetic perturbation analysis. The co-expression clustering analysis examines the top pathways of other well-studied genes that are highly co-expressed with our list of kinases. We utilized the co-expression clustering analyses deployed by the HPA and ARCHS web tools to associate predicted pathways with our targets. The HPA tissue and single-cell co-expression analysis for AATK and INSRR revealed that these protein kinases are co-expressed with genes that are functionally involved in central nervous system myelination, axon guidance, synaptic transmission, and microtubule cytoskeleton organization. Additionally, ARCHS shows central nervous system myelination (GO:0022010) as one of the top predicted pathways for AATK. Other predicted pathways for AATK include regulation of short-term neuronal synaptic plasticity (GO:0048172), synaptic vesicle maturation and exocytosis (GO:0016188, GO:0016079, and GO:2,000,300), and neuronal action potential propagation (GO:0019227 and GO:2,000,463). The predicted pathways for INSRR in ARCHS include Golgi transport vesicle coating (GO:0048200), insulin secretion (GO:0030073), and protein maturation by protein folding (GO:0022417). EPHA6 is part of the “Brain – Ion transport” cluster in the tissue expression clustering analysis in HPA and the “Neurons & Oligodendrocytes—Synaptic function” cluster in the single-cell co-expression analysis. These gene clusters also include other brain-specific pathways such as synaptic transmission, AMPA receptor activity, neuronal action potential, presynaptic membrane assembly, glutamate secretion, and regulation of NMDA receptor activity. ARCHS reveals similar pathways such as glutamate receptor signaling pathways (GO:0035235, GO:0007215, and GO:1,900,449) and additional pathways including synaptic plasticity (GO:0048172) and protein localization to synapse (GO:0035418). In HPA, LTK is a member of a cluster of genes that are involved in various immune response pathways such as adaptive and innate immune response, complement activation, and interferon-gamma-mediated signaling pathways and protein assembly and transport (Supplementary Table 6).
The second approach to associate functional pathways to our list of dark kinases is protein–protein interaction networks. STRING is a curated database of known and predicted protein–protein associations based on multiple resources including gene co-expression, experimental data, and text mining. We used STRING to extract the top 25 protein interactors with each of our list of kinases, using the default minimum required interaction score (medium confidence = 0.400) as the threshold. Next, we used the built-in functional network enrichment analysis in the STRING web tool. Using the previously described parameters, the AATK network had 24 interactors and only three enriched GO (Molecular Function) terms, including Ser-tRNA (Ala) hydrolase activity, alanine-tRNA ligase activity, and Rab guanyl-nucleotide exchange factor activity. EPHA6 network has 24 interactors and 317 enriched GO terms (Biological Process). The top enriched terms for the EPHA6 network include ephrin receptor signaling pathway, axon guidance, and neuron projection development and morphogenesis. The INSRR network contains 24 interactors and 252 enriched GO terms. The top enriched pathways include regulation of protein kinase B signaling, insulin receptor signaling pathway, phosphatidylinositol 3-kinase signaling, and MAPK cascade. There are 12 protein interactors in the LTK network and 85 enriched pathways. The top enriched terms include epidermal growth factor receptor signaling pathway, ERBB2 signaling pathway, generation of neurons, and insulin receptor signaling pathway. Finally, the TNK1 network has 12 interactors and only three enriched GO terms, including regulation of metalloendopeptidase and aspartic-type endopeptidase activity involved in amyloid precursor protein catabolic process (Supplementary Table 7).
To explore the results of our GO analyses, we performed “lookup” validation literature searches for select kinases and associated GO pathways (Supplementary Table 8). Notably, we found that the GO pathways axon guidance (GO:0007411) and innate immune response (GO:0045087) were associated with EPHA6 and TNK1 kinases, respectively. EPHA6 was highly expressed and localized to ganglion cells of the developing human retina. Patterns of EPHA6 expression in the macaque retina related to fovea development and ganglion cell projections suggest a role in the postnatal maintenance of neuronal projections [37]. A group investigating the impact of EPHA6 deletion on neuronal cell morphology in LacZ/LacZ mice discovered extensive impairments in the structure and function of both cells in the brain and spinal cord. These impairments include defects in memory and learning, changes in cellular morphology upon golgi staining at 2 months of age presenting aggregation of cells in the frontal cortical and mid-cortical regions [38]. Similarly, TNK1 was identified as a unique regulator of the ISG (interferon stimulated genes) pathway of the antiviral innate immune response in a high-throughput, genome wide cDNA screening assay used to identify genes regulating the ISG expression. Functionally, activated IFN-receptor complex recruits TNK1 from the cytoplasm, where upon phosphorylation it potentiates the JAK STAT signaling pathway. The authors noted there was a change in the phosphorylation state of STAT1 at two sites by western blot after 24 h of interferon beta exposure: tyrosine 701, and serine 727, however functional characterization of TNK1 activity was not conducted. Interestingly, the phosphosite containing tyrosine 701 maps to our medium affinity peptide logo (Supplementary Fig. 2E) corroborating the findings in our study [39].
The third approach to assess functional annotation of understudied kinases is with controlled genetic manipulation. The LINCS database hosts over a million transcriptional signatures of various cell lines with either genetic or chemical perturbations. We queried the iLINCS web portal to find and analyze gene knockdown or overexpression signatures of our list of dark kinases. Four of our five dark kinases have at least one gene knockdown signature, while TNK1 only has a gene overexpression signature. To keep the comparisons consistent, we selected the VCAP cell line as the primary cell line for the connectivity analysis because it was the only cell line that has gene knockdown signatures for all four kinases (AATK, EPHA6, INSRR, and LTK). To examine and functionally annotate the transcriptional “echo” of knocking down these kinases, we extracted the top differentially expressed genes (p-value < 0.05 and log2 fold change > 0.5 or < -0.5) from the LINCS knockdown signatures and performed gene set enrichment analysis using Enrichr. Using methods described in Sect. " Data analysis" of the dark kinome supplement, we selected top differentially expressed genes and extracted 99 genes from the AATK knockdown signature (LINCSKD_29946). Sorting by Enrichr’s Combined Score, the top associated pathways of this list of genes include regulation of astrocyte differentiation (GO:0048711, GO:0048710), regulation of insulin receptor signaling pathway (GO:0046627, GO:1,900,077), and regulation of extrinsic apoptotic signaling pathway via death domain receptors (GO:1,902,041). We extracted 113 differentially expressed genes from the EPHA6 knockdown signature (LINCSKD_30785). The top implicated pathways for these genes include protein insertion into mitochondrial membrane (GO:0001844, GO:0097345, and GO:0051204), regulation of inositol trisphosphate biosynthetic process (GO:0032960), and glutamate receptor signaling pathway (GO:0035235). For the INSRR knockdown signature (LINCSKD_31354), we extracted 98 differentially expressed genes and revealed pathways involved in regulation of hormone metabolic process (GO:0032352), sequestering of NF-kappaB (GO:0007253), and regulation of glial cell proliferation (GO:0060251). We extracted 100 differentially expressed genes from the LTK knockdown signature (LINCSKD_31563). The top implicated pathways for this list of genes include DNA ligation (GO:0006266), central nervous system projection neuron axonogenesis (GO:0021952), and acetyl-CoA metabolic process (GO:0006084). For the TNK1 overexpression signature (LINCSOE_9396), we extracted 278 differentially expressed genes and revealed pathways involved in regulation of protein import (GO:1,904,589), axo-dendritic transport (GO:0008088), and glycolytic process (GO:0006096) (Supplementary Table 8).
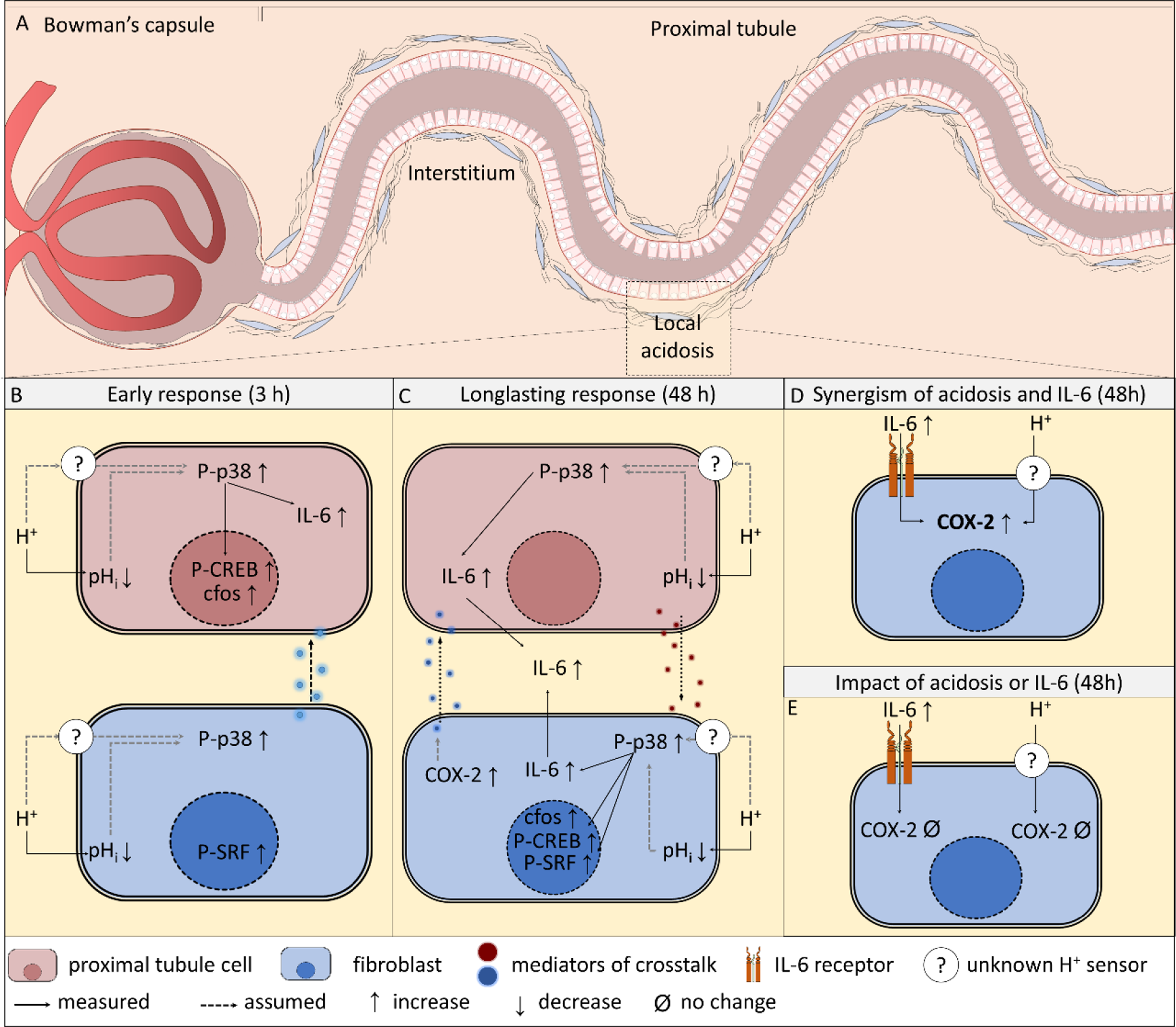
Connectivity analysis with psychiatric and neurogenerative diseasesTo elucidate the association of our list of dark kinases with diseases, more specifically with psychiatric and neurogenerative disorders, we performed a transcriptional connectivity analysis between gene perturbation signatures (from LINCS) and previously published disease signatures from schizophrenia (SCZ), major depressive disorder (MDD), and Alzheimer’s Disease (AD) datasets. Utilizing the disease signatures available in Kaleidoscope, we performed a Pearson correlation analysis to calculate the concordance scores between the LINCS gene perturbation and disease signatures using the top differentially expressed genes in each kinase knockdown or overexpression signature (Fig. 4A). Each node represents a transcriptional signature, and the edges are the Pearson correlation coefficients (Fig. 4B). Overall, the EPHA6 gene signature showed the most concordance with the disease groups, most notably with the SCZ and AD datasets. Some kinases showed relatively higher correlation with one disease group compared to the other diseases. For example, the AATK knockdown signature showed similarity with only the AD datasets. Additionally, we observed that the INSRR gene signature showed exclusively higher concordance with the SCZ datasets. The LTK knockdown signature showed the least amount of connectivity with the disease groups. The TNK1 overexpression signature also had low connectivity, especially with the SCZ and AD datasets.
Fig. 4
Transcriptional connectivity analysis explores concordance between AATK, EPHA6, TNK1, LTK, INSRR gene knockdown signatures and schizophrenia and Alzheimer’s Disease. A Pearson correlation analysis of LINCS knockdown signatures of AATK, EPHA6, INSRR, LTK, and TNK1 against 31 schizophrenia (SCZ), 21 Alzheimer’s Disease (AD), and 33 major depressive disorder (MDD) datasets extracted from the Kaleidoscope web application. The values represent correlation coefficients and color represent the sign of the coefficients (blue: positive correlation, red: negative correlation). The “*” within each node denotes the significance of the correlation analysis where the corrected p-value was under 0.05. B The connectivity analysis visualized as networks where each node represents a transcriptional signature and edges represent the correlation coefficients between signatures. The central node in each network represents the gene knockdown or overexpression signature of the corresponding kinase (AATK, EPHA6, INSRR, LTK, and TNK1) retrieved from iLINCS
Comments (0)