Chemicals and reagents
MG132 and JSH-23 were obtained from MedChemExpress. Cycloheximide (CHX) was sourced from Sigma-Aldrich, and recombinant HBP was procured from Novoprotein. Transfection reagents included Higene (Applygen Technology) and Lipo3000 (Life Technologies Corporation).
Cell culture treatment and transfection
HPMECs were acquired from Tongpai technology and cultured in Dulbecco's modifed Eagle's medium supplemented with 10% fetal bovine serum. After 2–3 days, they reached 80–90% confluence and were passaged using 0.25% trypsin. Cells were passaged at 80–90% confluence using 0.25% trypsin,and cells within 40 passages were used for all experiments. HPMECs were confirmed to be CD31-positive by immunofluorescence staining, ensuring the endothelial identity. In the cell experiments, the HBP group was treated with 1 µg/mL recombinant HBP protein for 24 h.
For transfection experiments, flag-HBP (MiaoLingBio, China),myc-TRIM21plasmids (MiaoLingBio, China), HA-Ub(MiaoLingBio, China), HA-Ub K48(MiaoLingBio, China), HA-Ub K63(MiaoLingBio, China) and corresponding empty vector were transfected into cells using transfection reagents according to the manufacturer's instructions. After 48 h of culture post-transfection, cells were uesd for subsequent experiments. TRIM21 gene silencing was performed in HPMECs by si-TRIM21(Haixing Biotech), with both siRNA and scrambled siRNA stock concentrations of 20 µM and working concentrations of 50–100 nM. Negative control (NC) included scrambled siRNA transfections. After 48 h, levels of TRIM21 proteins were detected by immunoblotting to verify transfection efficiency (sFigure 1). Subsequently, TRIM21 was knocked down using TRIM21 siRNA1.
Clinical data collection and human neutrophils isolation
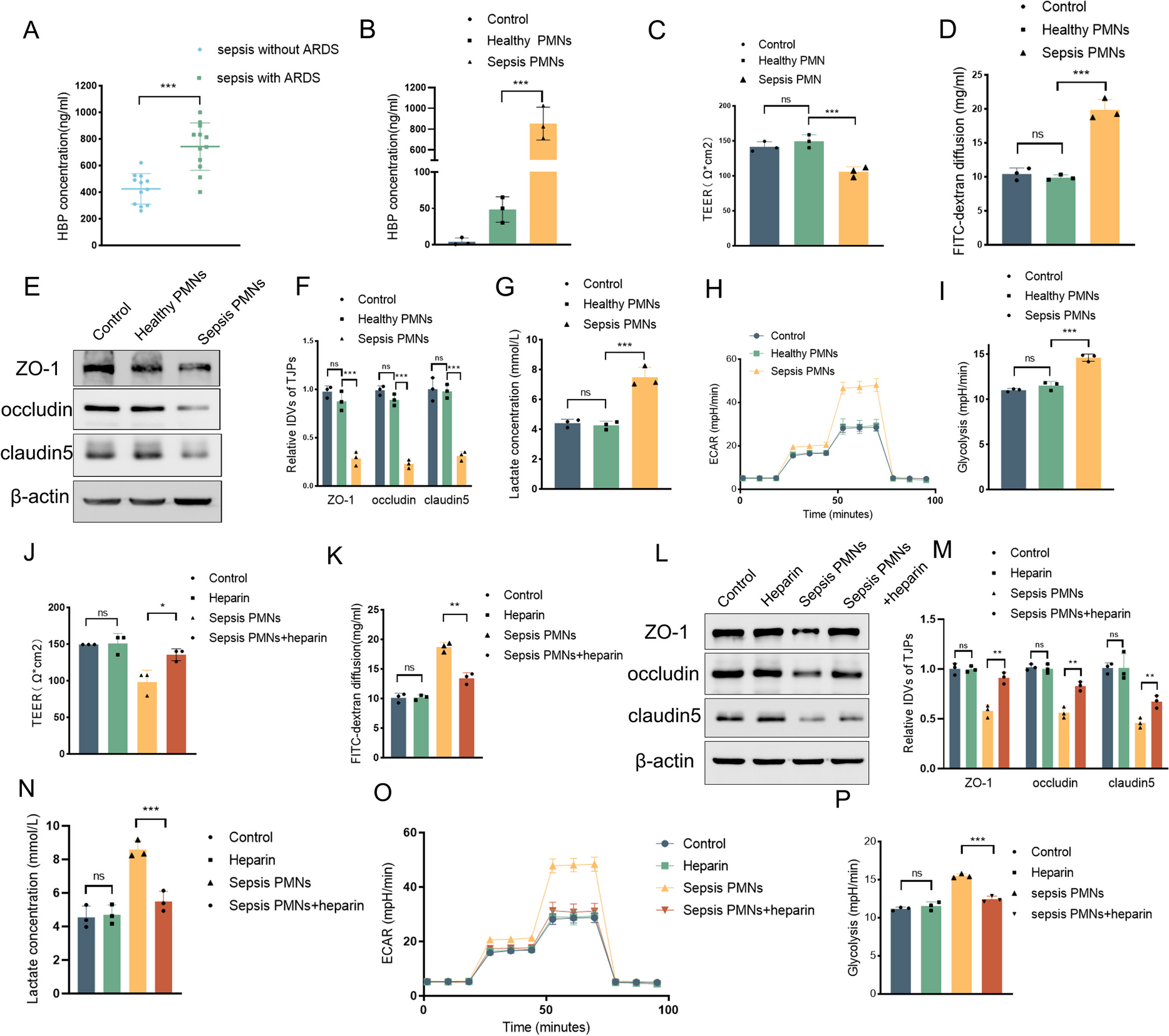
Clinical data on plasma HBP concentrations were collected from sepsis patients, both with and without ARDS, in the intensive care unit of the First Affiliated Hospital of China Medical University. Blood samples were collected in EDTA-coated tubes for neutrophil isolation isolation from healthy donors and sepsis patients who provided informed consent. All clinical data collection and experiments were conducted in accordance with the approval granted by the Ethics Committee of the First Affiliated Hospital of China Medical University (Approval Number: 2023–157). The inclusion criteria for participants are detailed in the Supplementary Materials.
Neutrophils were isolated using density gradient centrifugation by a commercial neutrophil extraction kit(P9040, Solarbio). Viability was assessed by Trypan Blue exclusion, which showed that more than 90% of the cells were viable. Cells were cultured in RPMI 1640 supplemented with 10% FBS.
Animal experiments and lung injury model
C57/BL male mice, aged 6–8 weeks, were obtained from Liaoning Changsheng Biological Company. The mice were housed under a clean environment with sufficient water and food, maintaining a normal circadian rhythm. Mice were anesthetized with isoflurane and euthanized by cervical dislocation at the end of the experiment. All procedures were approved by the Institutional Animal Care and Use Committee of China Medical University(Approval Number KT20240002).
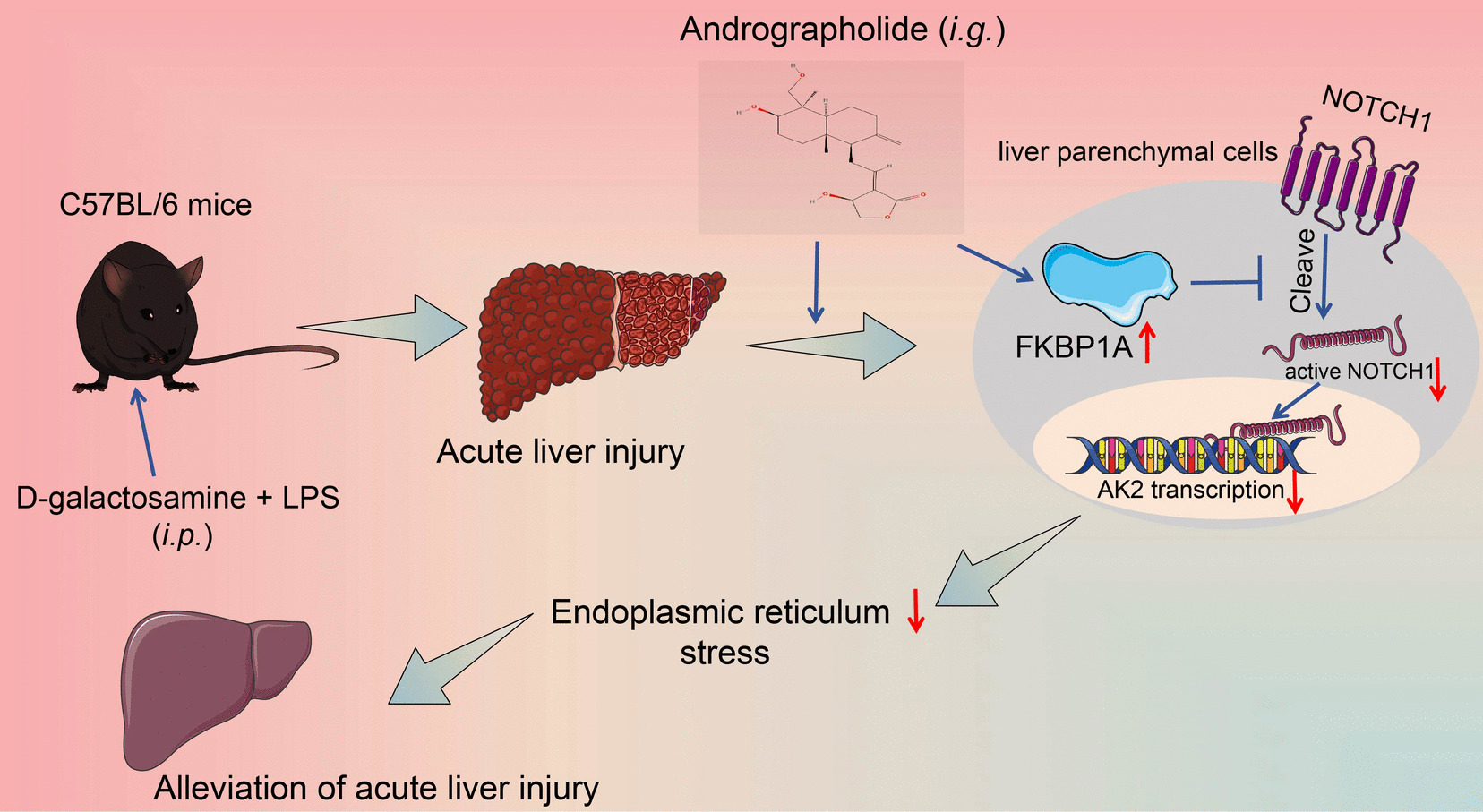
Based on preliminary experimental results, intravenous injection of 25 µg recombinant HBP protein was identified as the minimum dose required to induce lung injury in mice (sFig 2A, B). Therefore, this study employed 25 µg recombinant HBP administered via tail vein injection to establish the HBP-induced lung injury model. Mice were subjected to HBP treatment for 24 h (HBP group), while the control group received an equal volume of saline via tail vein injection. Each group consisted of five mice. To establish a LPS-induced acute lung injury model, mice were intratracheally administered 5 mg/kg LPS (2630, Sigma) in 50 µl saline for 24 h, with control mice receiving saline alone. Each group consisted of five mice.
Co-culture experiments
HPMECs and neutrophils were co-cultured in a Transwell system (0.4 µm pore size, 3413, Corning). HPMECs were seeded in the bottom chamber and neutrophils (5 × 10^5 cells) in the upper chamber. After 24 h, HPMEC samples were collected. To measure TEER and FITC-dextran leakage, HPMECs were seeded in the upper chambers and PMNs were placed in the lower chambers.
Enzyme-linked immunosorbent assay (Elisa)
HBP levels in EC supernatants were measured using a commercial ELISA kit (SEB461Hu, Cloud-Clone Corp, Wuhan, China). Samples were diluted 100-fold, and 100 µL of each was added to antibody-coated wells, followed by incubation at 37 °C for 90 min. After washing, 100 µL of biotin-labeled detection antibody was added and incubated at 37 °C for 60 min. Following another wash, 100 µL of HRP-avidin conjugate was added and incubated at 37 °C for 30 min. After a final wash, 90 µL of TMB substrate was added for color development at 37 °C in the dark for 15–20 min. The reaction was stopped with 50 µL of stop solution, and absorbance at 450 nm was measured using a microplate reader. HBP concentrations were calculated using a standard curve.
Western blot
Protein lysates were prepared using RIPA buffer, and protein concentrations were determined by BCA assay. Samples (20–40 µg protein) were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with the following primary antibodies: ZO-1 (21,773–1-AP, Proteintech, 1:5000), occludin (27,260–1-AP, Proteintech, 1:5000), claudin-5 (29,767–1-AP, Proteintech, 1:5000), TRIM21 (12,108–1-AP, Proteintech, 1:5000), P65 (80,979–1-RR, Proteintech, 1:1000), β-actin (81,115–1-RR, Proteintech, 1:10,000), Flag (66,008–4-Ig, Proteintech, 1:1000), Myc (16,286–1-AP, Proteintech, 1:5000), Histone 3 (17,168–1-AP, Proteintech, 1:10,000), HBP (MAB2200, R&D Systems, 1:1000), Ub (10,201–2-AP, Proteintech, 1:1000), K63-Ub (ab179434, Abcam, 1:1000), HA (51,064–2-AP, Proteintech, 1:10,000), ROCK2 (21,645–1-AP, Proteintech, 1:5000), ROCK1 (21,850–1-AP, Proteintech, 1:5000), Phospho-NF-κB p65 (Ser536) (80,379–2-RR, Proteintech, 1:1000), and Rho (mAb A4855, Abclonal, 1:1000). Gels were visualized using enhanced chemiluminescence.
Co-immunoprecipitation(Co-IP) and mass spectrometry analysis
Cells or lung tissue of mice were lysed in IP lysis (0.25% Sodium deoxycholate, 1 mM Tris–HCl 7.4, 0.5 M EDTA, 1% Triton-X 100, 1% NP40, 150 mM Nacl) containing protease inhibitors. After 30 min on ice, the lysate was centrifuged, and protein concentration was measured using a BCA assay. An aliquot of supernatant was set aside as input. The remaining lysate (1000 µg protein) was incubated with primary antibodies at 4 °C for 2 h, followed by overnight incubation with protein A/G magnetic beads (K1305, Apexbio, USA). The beads were then washed three times with IP buffer and proteins were boiled in 2 × SDS-PAGE buffer at 100 °C for 10 min.
The proteins were separated by SDS-PAGE and the relevant bands were excised, digested into peptides, and analyzed by LC–MS (PTM BIO). Data were processed with Proteome Discoverer 2.4 to identify protein interactions.
FITC-dextran leakage assay
Confluent layers of HPMECs in Transwell inserts were incubated with 1 mg/ml FITC-dextran to evaluate barrier permeability. After one hour of exposure, samples collected from the lower chamber were analyzed for fluorescence intensity to quantitatively assess the integrity of the endothelial barrier.
Transendothelial electrical resistance (TEER) measurement
TEER was assessed using a millicell-ERS instrument (Merck Millipore). ECs were cultured to confluence within Transwell inserts, and the culture medium was refreshed just prior to the measurements. Inserts were allowed to stabilize at 25 °C for 30 min before TEER values were recorded. During stabilization, the system was kept in a constant-temperature environment to ensure stable conditions. The inserts were undisturbed, allowing the cell layers and medium to equilibrate and eliminate any initial disturbances. This step minimized external influences and ensured a steady state before measurements. To account for background resistance, control values obtained from blank inserts were subtracted from those of the experimental groups. Subsequently, TEER values were normalized to the membrane surface area, resulting in measurements expressed as ohms*cm2. This normalization provides a quantitative assessment of endothelial barrier integrity.
Lactate measurement
Lactate concentrations in bronchoalveolar lavage fluid (BALF) and cell culture supernatants were determined using a Micro Lactate Assay Kit (KTB1100, Abbkine) in accordance with the manufacturer's instructions.
Extracellular acidification rate (ECAR)
ECAR was measured using an XF24 extracellular flux analyzer (Seahorse Bioscience). HPMECs at a density of 5 × 10^4 cells per well were plated on an XF24 plate. ECAR measurements were sequentially performed following the addition of glucose, oligomycin, and 2-deoxy-D-glucose (2-DG). Data analysis was conducted using Seahorse Wave software, which processed ECAR readings and evaluatedglycolysis capability following the manufacturer's protocol.
Immunofluorescence staining
Cells were cultured on glass slides, followed by fixation with 4% paraformaldehyde and permeabilization using 0.1% Triton X-100 for 10 min. To prevent non-specific binding, cells were blocked with 2% bovine serum albumin. Cells were incubated with primary antibodies against HBP (MAB461Hu24,Cloud-clone,1:100) and TRIM21(12,108–1-AP, proteintech,1:100) at 4 °C overnight. Subsequently, they were exposed to appropriate fluorescence-tagged secondary antibodies. Nuclei were stained with DAPI(G1012, servicebio) for 5 min. Fluorescence images were acquired using a Nikon A1 laser confocal microscope to evaluate protein localization.
Protein stability and degradation assay
To assess protein synthesis inhibition, HPMEC cells overexpressing HBP or a control plasmid were treated with 100 µg/mL CHX 24 h post-transfection. Total proteins were collected at different times for Western blot analysis. For protein degradation assay, cells were treated with 10 µM MG132, a proteasome inhibitor, or DMSO for the control group, and proteins were subsequently extracted for Western blotting to evaluate the effects on proteasomal degradation.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from HPMECs using a RNA extraction kit and quantified via spectrophotometry. Complementary DNA was synthesized using a reverse transcription master mix, and qPCR was performed with β-actin serving as an internal control. Primer information is detailed in Supplementary Table S1.
Lung wet-to-dry (W/D) ratio
Fresh right lung tissue was weighed and then dried at 68 °C until constant weight was achieved. Lung W/D ratio was calculated to assess pulmonary tissue exudation.
HE staining and lung inflammation score
Lung tissue was fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 µm thickness. Sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin for histological examination under a microscope. Lung injury was evaluated based on histopathological parameters including pulmonary edema, inflammation, hemorrhage, atelectasis, and hyaline membrane formation. Each parameter was scored semi-quantitatively from 0 to 4: 0 (no injury), 1 (< 25% involvement), 2 (25%−50%), 3 (50%−75%), and 4 (> 75%). The total injury score was the sum of all parameter scores. To reduce bias, 10 high-power fields were randomly selected for each animal, and scoring was performed independently by two investigators, with the average score representing the final lung injury score.
Evans blue leakage assay
Mice were injected intravenously with 0.5% Evans Blue dye. After one hour, pulmonary vasculature was cleared, and lung tissue was harvested and homogenized in formamide (1 mL per 100 mg tissue). Following incubation at 37 °C for 24 h and centrifugation, the supernatant's absorbance was measured to determine Evans Blue dye leakage, using a standard curve for quantification.
Isolation of primary pulmonary endothelial cells from mice
Primary pulmonary endothelial cells from mice were isolated using magnetic bead separation. Fresh lung tissue was first thoroughly washed with basal medium, then cut into small pieces and incubated in collagenase solution at 37 °C for 45 min. Next, 15µL of pre-incubated anti-CD31 antibody(553,370, BD)-conjugated magnetic beads(11,035, Thermo Fisher Scientific) were added per milliliter of cell suspension and incubated at 4 °C for 20 min. After discarding the cell suspension, the magnetic beads were washed with medium to obtain purified primary pulmonary endothelial cells.
Adenoviral vector infection
An adenoviral vector was purchased from Hanbio Tech (Shanghai, China). To knock down TRIM21 expression in the lung tissue of C57BL/6 mice, we employed HBAAV2/6-m-trim21 shRNA1-EGFP (shTRIM21), with HBAAV2/6-EGFP NC serving as the control vector. The vector, administered intratracheally at a dose of 5 × 10^10 viral genomes in 0.1 µL, achieved stable expression three weeks post-administration.
Wild-type mice without adenovirus transfection, mice infected with shNC adenovirus, and mice infected with shTRIM21 adenovirus were designated as WT, shNC, and shTRIM21 groups, respectively. Each group consisted of five mice. After receiving HBP or LPS treatment for the corresponding duration, the mice were sacrificed for subsequent experiments.
Statistical analysis
Data were analyzed using SPSS Version 23 and GraphPad Prism 9. Experimental results are reported as mean ± standard deviation (SD). For comparisons between two groups, the Student's t-test was applied. Differences among three or more groups were analyzed using one-way analysis of variance (ANOVA), with Bonferroni's method for inter-group comparisons. P < 0.05 was considered statistically significant.




Comments (0)