
Remember me

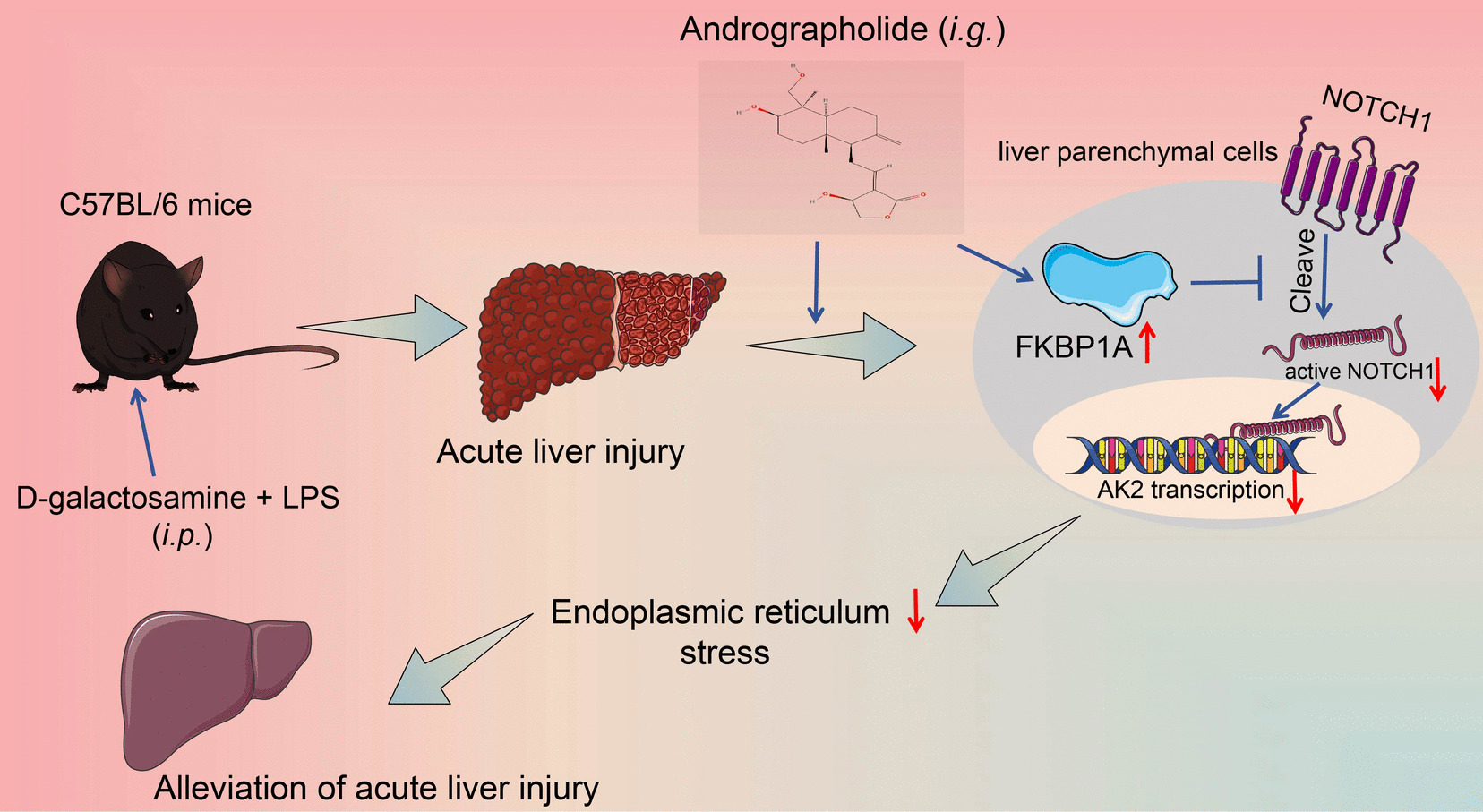


Human BMSCs (HUXMA-01001, Cyagen, USA) were maintained in DMEM medium (12,430,047, Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (12,483,020, Gibco, USA) and 1% penicillin/streptomycin (15,070,063, Gibco, USA) within a 37 °C incubator with 5% carbon dioxide. Cultivation of BMSCs in the third segment was carried out in DMEM encompassing 10% FBS until they achieved 80% confluence. To induce adipocytes, BMSCs underwent exposure to 1 µL/L dexamethasone (ST1254-50 mg, Beyotime, China), 0.5 µL/L isobutylmethylxanthine (410,957, Merck, USA), 60 µL/L indomethacin (SF6804-25 mg, Beyotime, China), and 10 µL/L insulin (P3376-100 IU, Beyotime, China). To promote osteogenesis, BMSCs were co-treated with 0.1 µL/L dexamethasone, 10 mmol/L glycerol 3-phosphate (G5251, Merck, USA), and 50 µL/L ascorbic acid (1,043,003, Merck, USA). After 3 weeks of induction, differentiation into adipocytes and osteoblasts was assessed using an Oil Red O staining kit (C0157S, Beyotime, China) and Alizarin Red staining (C0148S, Beyotime, China), respectively.
In the third passage, BMSCs were dissociated using 0.25% trypsin–EDTA (25,200,072, Gibco, USA) and enumerated. Nearly 1 × 106 cells were reconstituted in 200 µL of Fluorescence-activated cell sorting (FACS) buffer (660,585, BD Biosciences, USA). Afterward, the cells were chilled on ice and administered with 2 µL of fluorescent antibodies in conjunction with isotype controls, all throughout a period lasting 30 min. Following this, bone marrow stem cells (BMSCs) underwent rinsing with FACS solution, were then treated with 10% formalin (R04587, Merck, United States), and the proportion of positive antigens was assessed utilizing the BD FACSCalibur flow cytometer (BD Biosciences, USA). Antibodies used included CD90 (ab307736, 1:500), CD70 (ab77868, 1:500), and CD45 (ab10558, 1:1000) from Abcam, USA (Deng et al. 2021).
Isolation and recognition of BMSCs-EVsBMSCs were seeded in a 6-well plate and allowed to adhere for 24 h. The cells were rinsed with serum-free α-MEM (A1049001, Gibco, USA) and cultured in fresh serum-free α-MEM for another 24 h. Centrifugation at 2000 g led to the retrieval of the conditioned medium (CM). Ultracentrifugation was employed to isolate BMSCs-EVs in the CM. In brief, removal of cellular debris was achieved through centrifugation at 7000 × g for 20 min. Subsequently, microvesicles were pelleted at 16,500 × g over a period of 2 h at 4 °C. Following a 100,000 × g separation for 2 h at 4 °C, EVs underwent a rinse using PBS and underwent a subsequent centrifugation at 100,000 × g for an additional 2 h. GW4869 (HY-19363, MedChemexpress, USA), a commonly used neutral sphingomyelinase inhibitor at a concentration of 10 μM, was utilized to inhibit EVs generation and affect the composition and biological activities of EVs membranes (Andreeff et al. 1987; Dorronsoro et al. 2021).
Transmission electron microscopy (TEM) was employed to identify EVs: A copper grid was utilized to receive 20 μL of EVs, which were allowed to settle for 3 min, after which excess liquid was removed using filter paper. Subsequently, 30 μL of phosphotungstic acid solution (pH 6.8) (79,690, Merck, USA) was added. Subsequently, the grid underwent air-drying subsequent to a 5-min exposure at ambient temperature, leading to examination using a transmission electron microscope (JEOL's JEM-1011 model based in Tokyo, Japan) with an accelerating voltage set at 80 kV, and visuals were obtained via a side-mounted Camera-Megaview III (Soft Imaging System, Muenster, Germany).
The size and quantity of EVs were measured through the use of nanoparticle tracking analysis (NTA): Suspended in PBS, the EVs samples were diluted by a factor of 500 using Milli-Q water and were carefully introduced into the sample space of a NanoSight LM10 tool from the UK's Malvern facility with the aid of a sterile syringe, ensuring the absence of bubbles until the space was fully occupied. Utilization of NanoSight software 2.3 from Malvern, UK, was crucial during the video analysis procedure. Specifically, a gain setting of 6.0 and a threshold value of 11 facilitated the monitoring of particle movement, leading to the generation of profiles depicting concentration and size distribution for the diluted samples. Consequently, the calculation of the EVs' initial concentration was made based on the dilution factor.
The surface markers of EVs were identified using Western blot (WB): Two sample groups were established: the GW4869 group (where EV activity was inhibited using GW4869 as a control group) and the BMSCs-EVs group (used to verify EV extraction purity). EVs suspended in RIPA lysis buffer (89,901, Thermo, USA) were probed for specific markers such as CD9, CD63, TSG101, and HSP70, with Calnexin serving as a negative control, utilizing rabbit anti-CD9 (ab307085, 1:1000), anti-CD63 (ab134045, 1:1000), anti-TSG101 (ab125011, 1:1000), anti-HSP70 (ab2787, 1:1000), and anti-Calnexin (ab22595, 1:1000) antibodies from abcam (UK) (Li et al. 2021; He et al. 2020; Gangadaran et al. 2017; Zhou et al. 2019). Every trial was conducted thrice.
Silence and Overexpression of B4GALT3 through Lentivirus Construction and Transfection.
Initially, formulated were three different sets of sequences to target B4GALT3, where a sequence without any interfering elements was utilized as a control (sh-NC). Table S1 contains the primer sequences, and GenePharma® from Shanghai, China, synthesized the oligonucleotides. Utilization of the lentivirus packaging system pLKO.1 involved the co-transfection of the packaged virus and target vector into 293 T cells (H4-1601, Cyagen, USA) using Lipofectamine 2000 (Invitrogen, USA) once the cellular confluence reached 80–90%. Upon completing a 2-day cell incubation, the fluid medium was procured, subjected to centrifugal force, and detected to harbor viral elements. The viruses proliferating exponentially were collected, and the viral potency was ascertained. Manufactured and packaged by Genechem located in Shanghai, China, the lentiviral vector designated LV-B4GALT3 was tailored for the amplification of B4GALT3 expression. After entering the logarithmic stage, the cells underwent digestion and transfer using trypsin, leading to a concentration of 5 × 104 cells/mL of BMSCs, which were subsequently placed in a 6-well tray, containing 2 mL per well. Each lentivirus (MOI = 10, viral titer of 1 × 108 TU/mL) was introduced into the cellular medium and cultured for a period of 48 h. The process of selecting stable cell lines involved the use of 2 µg/mL puromycin (UC0E03, Sigma-Aldrich, Germany) for a period of 14 days (Zhang et al. 2019; Wang et al. 2020; Ni et al. 2021; Shi et al. 2020).
Grouping of BMSCs-EVs Co-culture with CAFs cellsHuman CAFs cells (SNP-H067, SUNNCELL) were co-cultured with BMSCs-EVs for 24 and 48 h in DMEM medium containing 10% FBS and 1% penicillin/streptomycin. The co-cultures were incubated at 37 °C with 5% CO2. The co-culture experiments of EVs with CAFs cells were divided into 6 groups, with detailed procedures outlined in Table S2. CAFs cells were treated with 0.5 μM of the NF-κB activator Diprovocim (TNFα; HY-123942, MedChemexpress, USA) for 24 h (Chen et al. 2017; Peng et al. 2023).
Observation of the uptake of EVs by CAFs using fluorescence microscopyCAFs were pre-seeded onto glass coverslips in a 24-well plate. Each well was filled with a cell suspension composed of 5 × 104 cells per well. Upon the adherence of CAFs, pre-stained EVs supernatant was added, and co-cultured for 24 h. Dil dye (C1036, Beyotime, China) was mixed with 40 μg of EVs to reach a last level of 25 μM. Subsequent to a 30-min room temperature incubation, the undyed molecules were separated by swift centrifugation. Subsequently, the cells received three PBS washes and were subsequently immobilized with 4% paraformaldehyde (PFA, AR1068, Boster, China) for 30 min. Ultimately, the staining of cell nuclei with DAPI (C1005, Beyotime, China) lasted 30 min, and CAFs were visualized at × 400 magnification applying a fluorescence microscope equipped with a camera (BX53, Olympus, Japan). Blue fluorescence indicated the CAFs nuclei, while red fluorescence indicated Dil. Results were presented as the ratio of Dil fluorescence area to DAPI fluorescence area, computed utilizing ImageJ software developed by the National Institutes of Health in the United States. Examination of each cell involved 5 image sections and the observation of 6–10 fields chosen at random (Cui et al. 2021; Xiong et al. 2022). The experiment was replicated on three occasions.
Grouping of CAFs and HCC Cell Co-culture experimentsCo-culture of CAFs with HCC Cells: Normal liver cell line THLE-2 (MZ-8312) and liver cancer cell lines Huh-7 (MZ-0095), SNU-182 (MZ-2656), and JHH7 (MZ-2707) were all obtained from Ningbo Mingzhou Biotechnology Co., Ltd. THLE-2 cellular cultures were grown in 1640 growth medium (R8758, Sigma-Aldrich, USA) comprising 10% FBS, while Huh-7, SNU-182, and JHH7 cells were sustained in DMEM with 10% FBS and 1% penicillin/streptomycin.
CAFs previously incubated with PBS or BMSCs-EVs were seeded in the upper chamber of Transwell co-culture systems (354,480, Corning, USA), with pores measuring 0.4 µm and a cell density of 1 × 105 cells/ml, while the lower enclosure served as the setting for seeding HCC cells. Co-cultivation of CAFs and HCC cells occurred over a period of 24 h prior to the commencement of subsequent experiments involving HCC cells.
Based on experimental requirements, HCC cells co-cultured with CAFs treated differently were divided into the following six groups: PBS, EVs, EVslv−NC, EVslv−B4GALT3, EVslv−NC+TNFα, and EVslv−B4GALT3 + TNFα. In the PBS, EVs, EVslv−NC, EVslv−B4GALT3, EVslv−NC + TNFα, and EVslv−B4GALT3 + TNFα groups, CAFs were treated with PBS, EVs, EVslv−NC, EVslv−B4GALT3, EVslv−NC + TNFα, and EVslv−B4GALT3 + TNFα, respectively, before co-culturing with HCC cells (Huh-7, SNU-182, or JHH7) (Salah et al. 2022; Zhao et al. 2022).
Construction and grouping of HCC mouse modelsThe study involved the acquisition of fifty-six vigorous male BALB/c nude mice, aged 4 to 6 weeks, from Charles River, China. Each mouse was accommodated in isolation within SPF-certified animal quarters, with humidity levels set at 60–65% and temperature precisely adjusted to fall between 22 and 25 ℃. Abundant nourishment and hydration were supplied to the mice within a 12-h light–dark rhythm. The experiment initiated after a week of adaptation, with the mice's health being evaluated in advance. The wellbeing of each living organism was ensured in compliance with the guidelines outlined in the "Guide for the Care and Use of Laboratory Animals" published by the National Academy of Sciences and endorsed by the National Institutes of Health. Authorization for the experimental procedures and animal care protocols was provided by the Institutional Animal Ethics Committee (Ethics Approval No: [2022]298).
For the establishment of the orthotopic HCC model, introduction of the orthotopic HCC model involved the injection of 30 μl PBS mixed with 2 × 106 Huh-7 cells into the liver lobes of mice. Starting from the 7th-day post-tumor cell injection, injections were administered to mice intravenously with 100 μl of PBS carrying EVs (10 μg per mouse) or without EVs every second day. After 4 weeks of inoculation, mice were euthanized with 100 mg/kg of pentobarbital sodium (11,715, Merck, USA), and their livers were harvested. THE establishment of the HCC lung metastasis model was accomplished by injecting 2 × 106 Huh-7 cells diluted in 100 μl of PBS into the mice's blood circulation via the tail vein. Starting from the 5th-day post-tumor cell injection, administration of 100 μl of PBS, including EVs (10 μg per rodent), or without EVs, was done through intravenous injection to the mice at four-day intervals. Mice were euthanized 8 weeks post-tumor cell injection, and their lungs were excised. Tumors were dissected to measure the maximum diameter and weight (Jiang et al. 2020; Hou et al. 2020).
Mice were arbitrarily separated into categories in the subsequent manner: Control group (5 mice) and HCC group (51 mice). The Control group received no treatment. The HCC group was further divided into subgroups: HCC group (5 mice), PBS group (3 mice), EVs group (3 mice), HCC + PBS group (10 mice), HCC + EVslv−NC group (10 mice), HCC + EVslv−B4GALT3 group (10 mice), and HCC + EVslv−B4GALT3 + TNFα group (10 mice).
In the HCC group, Huh-7 cells were injected to establish an in situ HCC model. In the PBS group and EVs group, after establishing the in situ HCC model, mice received tail vein injections of PBS or BMSCs-EVs respectively. Liver tissues of mice from the Control group, HCC group, PBS group, and EVs group were collected for subsequent transcriptomic sequencing (Jiang et al. 2020; Hou et al. 2020).
For the HCC + PBS group, HCC + EVslv-NC group, HCC + EVslv-B4GALT3 group, and HCC + EVslv-B4GALT3 + TNFα group, CAFs treated with PBS, EVslv-NC, EVslv-B4GALT3, or EVslv-B4GALT3 + TNFα were mixed with luciferase-labeled Huh-7 cells in a 1:1 ratio and suspended in PBS, then injected into the liver lobes of mice or tail veins to establish in situ HCC models and lung metastasis HCC models. Five HCC mice from each group were used for in situ or lung metastasis model formation. Following 4 or 8 weeks of model establishment, tumor burden was detected using IVIS Lumina III (PerkinElmer) for bioluminescence imaging. Subsequently, liver or lung tissues were collected from the mice (Jiang et al. 2020; Hou et al. 2020; Liu et al. 2021).
Histological examinationMouse liver, tumor tissue, and lung tissue were stained using an H&E staining kit (C0105, Beyotime, China). The following are the exact measures taken in progression: the liver and lung samples stemming from the murine models were initially placed in 10% neutral buffered formalin at a temperature of 4 °C for a complete period of 24 h. Following this, they underwent dehydration, were coated with paraffin, and were sliced into individual sections. Xylene was used to deparaffinize the sections, followed by gradual alcohol hydration and rinsing with distilled water. Afterward, the items were immersed in hematoxylin solution for 5–10 min, any extra dye was cleared off with deionized water for about 10 min, and later treated with eosin solution for a period spanning from 30 s to 2 min. Finally, they underwent gradient alcohol dehydration and xylene clearing. Neutral resin or an alternate mounting medium was used to fix the sections, which were then inspected and photographed under an inverted microscope (IX73, Olympus, Japan) (Bai et al. 2022).
TUNEL stainingThe TUNEL Kit reagent (C1086, Beyotime, China) was utilized to assess the apoptosis status in mouse tumor tissues. Tissue slices were treated with 3% H2O2 and then incubated with 50 μL TUNEL for 60 min, avoiding exposure to light at 37 °C. DAPI staining was applied to the cell nuclei, allowing for a duration of 30 min, after which they were rinsed thrice with PBS. Subsequently, a fluorescence microscope with a camera (BX53, Olympus, Japan) was employed to photograph tumor tissues at a × 400 magnification. Calculation of the TUNEL fluorescence area with respect to the DAPI fluorescence area was conducted via the ImageJ application. Every collection was composed of 5 mice, encompassing 5 parts for each mouse randomly viewed in 6–10 territories. ImageJ tool was applied for the assessment of TUNEL-positive cell proportion (Tang et al. 2023).
Immunohistochemistry (IHC) experimentTumor tissues were processed through the following steps: fixation in 4% PFA, dehydration, clearing, paraffin embedding, and ultimately sectioning of the tissues. For IHC staining, the sections underwent deparaffinization and rehydration, followed by antigen retrieval. The sections were then immersed in a vessel with PBS and subjected to microwave heating until boiling occurred. Subsequently, immunostaining was carried out applying the universal two-step detection kit (PV-9000, ProteinTech, USA) abiding by the manufacturer's prescribed protocol. The main antibodies utilized were as outlined: Ki67 (12,335–1-AP, 1:500), NF-κB p-p65 (ab131100, 1:100), N-cadherin (ab76011, 1:500), E-cadherin (ab76319, 1:500), and Vimentin (ab92547, 1:500), showing significantly increased expression levels. The utilization of an optical microscope (CX43, OLYMPUS, Japan) facilitated the observation and recording of staining outcomes. Staining in positive regions appeared as light brown or tan hue. Every collection was made up of 5 mice, having 5 segments for every mouse, procured from the matching tissue sample for every mouse. Different protein staining was performed on each Sect. 6–10 fields were randomly selected for observation, and the amount of positive cells was documented. Inspection was executed utilizing the image interpreting system (Aperio Scanscope System, Vista, CA) (Cai et al. 2021).
HCC Mice liver tissue transcriptomic investigationMice in the Control, HCC, PBS, and EVs groups provided liver tissue samples for analysis. The Invitrogen Total RNA Isolation Reagent Kit (12,183,555, USA) was utilized for the isolation and extraction of total RNA from the samples, followed by quantification through OD value assessment. Agarose gel electrophoresis was employed to evaluate the integrity of the total RNA, with a RIN value between 7 and 10 considered high quality. High-quality total RNA underwent reverse transcription to generate cDNA, followed by the assembly of libraries and sequencing conducted on Illumina's NextSeq 500 platform. Initial reads were produced from the sequencing's raw image data by converting them using base calling techniques. Ensuring the superior quality of primary documents, cutadapt was invoked to excise the adapter sequences used for sequencing and eliminate low-quality sequences, resulting in the remaining material designated as "purified reads." After aligning these sequences with the mouse reference genome via the application Hisat2, the quantitative examination of gene expression was performed by employing the R software's limma package, ultimately creating a matrix displaying gene expression levels (Li et al. 2022a, b).
Bioinformatics AnalysisThe "limma" software in the R linguistic framework was employed to examine the variations in mRNA expression levels identified in the transcriptome sequencing information. The thresholds applied to differentiate the mRNA distinctions between the Control and HCC samples were determined by ∣Log2 Fold change∣ > 2 and P.adj < 0.05. For the PBS and EVs groups, the criteria were set at ∣Log2 Fold change∣ > 1 and P.adj < 0.05. Implementation of the ggplot2 software led to the creation of volcano sketches, and the pheatmap package in R facilitated heatmap generation. The Exome Academic Database was employed for the formulation of Venn diagrams (Langfelder and Horvath 2008). The gene set for CAFs was retrieved and downloaded from the GeneCards database (https://www.genecards.org/). The Jaspar database was utilized to predict the binding positions of B4GALT3 with the NF-κB signaling pathway (https://jaspar.genereg.net/). Conducting GO and KEGG pathway assessments in R language was enabled by the integration of "clusterProfiler," "org.Hs.eg.db," "enrichplot," "DOSE," and "ggplot2" packages (Liang et al. 2022, Du et al. 2022).
Immunofluorescent (IF) stainingAfter fixation in 4% PFA for a quarter of an hour, both cells and tumor tissues underwent dual PBS rinses and were subsequently made penetrable with 0.5% Triton X-100 (P0096, Belltime, China) for 10 min. Later on, both tissues and cells were subjected to overnight incubation at 4 °C with primary antibodies against rabbit anti-α-SMA (ab7817, 1:100), FAP (ab314456, 1:100), NF-κB p-p65 (ab131100, 1:100), B4GALT3 (20,330–1-AP, 1:100), Collagen I (ab270993, 1:100), NF-κB p65 (ab16502, 1:100). Post triple PBS washes, tissue sections were treated with secondary antibodies conjugated with Alexa Fluor 647 (ab150083, 1:200) or Alexa Fluor 488 (ab150077, 1:200) for an hour. Afterward, the segments were rinsed thrice with PBS solution and subjected to staining with DAPI (C1005, Beyotime, 10 μg/mL) for a period of 10 min. The sections were maintained in cold storage at 4 °C and inspected through the use of a fluorescence microscope (IMT-2, Olympus, Japan). For cellular experiments, each cell was evaluated on 5 slices, observing 6–10 fields randomly, repeated 3 times per experiment; for animal experiments, each group consisted of 5 mice, each subjected to 5 slices, with 6–10 fields observed randomly. ImageJ software facilitated the measurement of the fluorescence area of α-SMA, FAP, NF-κB p-p65, and B4GALT3 concerning DAPI fluorescence area, or it was utilized to assess the fluorescence intensity of NF-κB p65 co-localized with DAPI within the cell population. All antibodies were purchased from abcam, except B4GALT3 from Protein tech (Yue et al. 2019).
Dual luciferase reporter gene experimentFrom the NCBI database, the protein sequence for B4GALT3 and the DNA sequence of the NF-κB promoter were extracted and processed for molecular docking analysis using the HDOCK website (http://hdock.phys.hust.edu.cn/). The most stable binding structure was identified as Model_1 (Figure S1A). The pdb file of Model_1 was subsequently uploaded to the PDBsum website (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) for further analysis of the binding sequence information between B4GALT3 and the NF-κB promoter DNA (Figure S1B). Co-transfecting BMSCs with lv-NC and lv-B4GALT3 involved the delivery of a fluorescent luciferase reporter plasmid harboring the NF-κB promoter sequence (TATATCTGGC) through the Lipofectamine 2000 reagent kit (11,668,019, Thermo, USA). To serve as an internal control, the cell's luciferase activity post-transfection for 48 h was assessed using the Renilla luciferase assay kit (K801-200, Biovision, USA). Employing the Dual-Luciferase Reporter Assay System from Promega in Madison, WI, USA, allowed for the detection of the luciferase reporter gene. Calculating the ratio between Firefly luciferase assay values (RLU) and Renilla luciferase assay values (RLU) enabled the determination of the activation level of the specific reporter gene (Taniue et al. 2016). Each experiment was repeated thrice for accuracy.
Mutant vectors of dual-luciferase reporter gene constructsSubsequently, the wild-type NF-κB (NF-κB-WT) site (5'-TATATCTGGC-3') and the mutant NF-κB (NF-κB-MUT) site (5'-CGCGAGTTCC-3') were individually inserted into the pGL-3 luciferase reporter vector (4,351,372, Thermo Fisher, USA). The co-transfection of NF-κB-WT and NF-κB-MUT luciferase reporter plasmids with lentiviral vectors carrying lv-NC and lv-B4GALT3 into CAFs was followed by cell retrieval and lysis after 48 h. The supernatant, derived post-centrifugation at 250 × g for 3–5 min, was integrated with the Dual-Luciferase® Reporter Assay System (E1910a, Promega, USA) for the evaluation of luciferase performance. Treatment of individual cell samples involved the application of 100 μL of Firefly Luciferase working solution for Firefly Luciferase detection and 100 μL of Renilla Luciferase working solution for Renilla Luciferase detection, comparing the relative luciferase activity of Firefly to Renilla Luciferase (Jin et al. 2020). Each experiment was repeated thrice.
Scratching experimentIn the cell scratching experiment, the 6-well plate received HCC cells at full seeding density. The cell layer was scratched using the tip of a sterile pipette (200 μL) and then gently washed with PBS solution three times. Cell observation and photography were conducted at 0, 24, and 48 h intervals to analyze the healing area of the scratch in each group. Before the scratching study, cells were treated with cytochalasin D (1 μg/mL, M5353, Sigma-Aldrich) for 1 h to eliminate any cell proliferation defects. The scratch's width was gauged initially (T = 0 h) and subsequently at 24 h (T = 24 h) and 48 h (T = 48 h). Cell migration ability was evaluated by calculating the difference in scratch width between the two time points and expressed as a percentage. Five images were taken for each cell, and inspections took place in 6–10 fields of sight that were chosen randomly (Song et al. 2015). Thrice the test was replicated for validation purposes.
Transwell experimentTwo types of cell culture inserts were utilized in the Transwell experiment: Application of Transwell PC filter cell incubation equipment (CLS3422, Corning, USA) and BioCoat Matrigel infiltration assemblies employing 8.0 μm PET layer (354,480, Corning, USA). Co-cultured HCC cells with CAFs were seeded at 1 × 104 cells in the upper chamber with or without matrix gel, followed by adding DMEM containing 10% FBS to the lower chamber. After incubation for 24 h post-seeding, cells underwent fixation with 4% PFA followed by a gentle removal from the upper polycarbonate membrane employing a cotton swab. A crystal violet stain solution (C0121, Beyotime, China) was used to color cells that underwent migration and invasion. Examination under an inverted microscope (XDS-900, Caikon, China) revealed five partitions per cell in 6–10 randomly selected fields. The "Analyze Particles" feature in ImageJ was employed to quantify the invading cell numbers in every single image (Mao et al. 2021a, b). The validation process included three separate repetitions for every experiment.
Detection of cell apoptosis by flow cytometryThe dual staining approach utilizing Annexin V-FITC and propidium iodide (PI) was employed to analyze cellular apoptosis in Huh-7 cells. 2 × 105 cells of the Huh-7 cell line were placed in each well of a 6-well plate and exposed to trypsin (R001100, Gibco, USA). Retrieval of the supernatant was achieved through centrifugation of cells at 800 g in 15 mL centrifuge tubes. Suspension of the pellet took place in 500 μL of binding buffer, followed by a 15-min incubation period with 5 μL of FITC and 5 μL of PI in a darkened environment, in line with the protocols specified by the apoptosis detection kit (556,547, BD Biosciences, USA). A flow cytometer obtained from BD Biosciences in the USA was employed to analyze apoptosis in the cell suspension. Necrotic cells were observed in the quadrant positioned at the top left, late apoptotic cells in the quadrant positioned at the top right, early apoptotic cells in the quadrant positioned at the bottom right, and normal cells in the quadrant positioned at the bottom left. The calculation of the apoptosis rate involved summing the percentages of cells in the upper right and lower right quadrants (Zuo et al. 2022). Every trial was repeated on three occasions.
RT-qPCRIsolation of total RNA from cellular populations and tumor samples was performed applying TRIzol (15,596,026, ThermoFisher, USA), followed by the determination of its concentration and purity employing a nanodrop2000 spectrophotometer (ThermoFisher, USA). The extracted RNA was reverse-transcribed to cDNA following the instructions of the PrimeScript RT reagent Kit (RR047A, Takara, Japan) and then subjected to RT-PCR applying the Fast SYBR Green PCR Master Mix (11,736,059, ThermoFisher, USA). The internal control chosen was GAPDH, with triplicate setups for each well. Assessment of gene expression levels was done employing the 2−ΔΔCt technique, and the trial was reiterated three times. Table S3 contains the primer sequences applied for RT-qPCR in this investigation (Liang et al. 2017).
WBLysis of cells and tumor tissues was conducted in RIPA lysis buffer (P0013B, Beyotime Biotechnology, China), with the quantification of protein concentrations accomplished using the BCA method (A53226, Thermo Fisher Scientific, USA). Separation of the proteins was carried out via polyacrylamide gel electrophoresis, and subsequently, they were transferred onto a PVDF membrane (PVH85R, Millipore, Germany) applying a wet transfer method. Blocking of the membrane occurred using 5% BSA for an hour at room temperature, followed by an incubation overnight at 4 °C with the primary antibodies mentioned: rabbit anti-α-SMA (ab7817, 1:1000), FAP (ab314456, 1:1000), B4GALT3 (20,330–1-AP, 1:1000), rabbit anti-Collagen I (ab316222, 1:1000), rabbit anti-p-IκBα (2859, 1:1000), rabbit anti-IκBα (4812, 1:1000), rabbit anti-NF-κB p65 (ab32536, 1:1000), rabbit anti-NF-κB p-p65 (ab131100, 1:1000), and rabbit anti-GAPDH (ab8245, 1:1000). The cleansing of the membrane was followed by incubation with a secondary antibody that was conjugated with HRP-goat anti-rabbit IgG (ab6721, at a concentration of 1:5000) for up to 2 h. Three 5-min cycles of TBST washings were conducted on the membrane. The identification of proteins was executed employing a chemiluminescent imaging device, and the evaluation of protein quantities was carried out utilizing ImageJ software, assessing the protein expression levels against the grayscale proportion of the loading control GAPDH or the corresponding non-phosphorylated protein. Every test underwent replication on three occasions. Notably, B4GALT3 was procured from Protein tech, p-IκBα and IκBα were obtained from Cell Signaling Technology, and all other antibodies were purchased from Abcam (Zou et al. 2016).
Statistical analysis in researchUtilization of SPSS software (version 21.0, IBM, USA) facilitated the statistical analysis of data in this study, presenting all data as mean ± standard deviation (mean ± SD). All significance tests were two-tailed. Initially, the normal distribution was assessed using the Kolmogorov–Smirnov test. Assuming normal data distribution, two groups were compared using unpaired t-tests, with multiple group comparisons conducted through one-way ANOVA or repeated measures ANOVA. The Wilcoxon signed-rank test was utilized to examine significant disparities between the dual groups for data that was not distributed normally. Significance in statistics was denoted by a p-value lower than 0.05 (Wan et al. 2023; Xu et al. 2022).
Comments (0)