
Remember me
Sepsis, a systemic inflammation syndrome with high occurrence and mortality rates, results in tissue and organ damage due to an uncontrolled response to infection.[1] The kidney is a primary organ affected in the early stages of sepsis. Approximately two-thirds of patients experiencing septic shock also suffer from acute kidney injury (AKI),[2] and half of these patients develop AKI before arriving at the emergency department.[3] Therefore, it is generally considered that AKI can serve as an early indicator of sepsis. Approximately 20% of patients with AKI will develop chronic kidney disease or progress to end-stage renal disease.[3] Sepsis-induced AKI is characterized by oxidative stress, inflammatory responses, renal tubular epithelial cell apoptosis, and abnormalities in kidney hemodynamics.[4,5] In particular, the disruption of inflammatory responses leads to an increase in inflammatory cytokines, which further trigger cell apoptosis in the kidney.[6] Therefore, studying the above biological manifestations and potential interventions may contribute to the development of therapeutic methods for sepsis-induced AKI.
Annexins, a widely studied family of genes, can bind to phospholipids in a manner that depends on the presence of Ca2+.[7] They participate in numerous physiological processes, including but not limited to preventing blood clots, reducing inflammation, facilitating cellular uptake and release, transmitting signals within cells, promoting cell growth and differentiation, and regulating apoptosis.[8] Annexin A3 (ANXA3), an important member of the annexin family, is located on chromosome 4q13–q22 and codes for two protein isoforms.[9] Its functions include cytoskeletal protein interactions, cell differentiation, and inflammatory response regulation. ANXA3 dysregulation has been linked to various human diseases. For example, the high expression of ANXA3 mediates tumor growth, apoptosis, and metastasis during the development of breast,[10] pancreatic,[11] and bladder[12] cancers. Furthermore, Wang et al. identified ANXA3 as a novel biomarker to distinguish pediatric patients with sepsis from healthy controls.[13] Importantly, Liang et al. reported that the depletion of ANXA3 attenuates inflammatory responses and cell apoptosis in sepsis-associated acute lung injury, another deadly complication of sepsis.[14] However, the involvement of ANXA3 in sepsis-related AKI is currently at an early stage of investigation.
In this work, we preliminarily uncovered the pathogenic role of ANXA3 in patients with AKI and sepsis. Moreover, we found that knocking down ANXA3 mitigates sepsis-induced AKI through regulating endoplasmic reticulum (ER) and oxidative stresses, apoptosis, and inflammation in vitro. These discoveries could provide insight into the underlying function of ANXA3 in the development of sepsis-induced AKI and present possible treatment options for individuals with sepsis-induced AKI.
MATERIAL AND METHODS Serum samplesA total of 20 healthy individuals (age: 45.2 ± 8.9 years; male: female ratio of 10: 10) and 20 patients with sepsis and AKI (age: 44.7 ± 9.5 years; male: female ratio of 10:10) were recruited from our hospital. Blood samples were collected from the participants. The inclusion criteria for patients are as follows: Body temperature (<36°C or >38.5°C), heart rate (>90 beats/min), tachypnea (>20 breaths/min or PaCO2 <32 mmHg), leukopenia (<4000/mm3), and leukocytosis (>12000/mm3). The exclusion criteria were the presence of malignancy, pregnancy in female patients, chronic renal insufficiency, the use of nephrotoxic drugs, or the reception of any kind of renal replacement therapy. Serum samples were centrifuged and stored at −80°C until utilized. This study was conducted by following the ethical standards established by our hospital’s ethics committee (approval no. 2020ky327). All participants provided informed consent, and this research complied with the principles stated in the Declaration of Helsinki.
AKI cell model establishmentProcell (Wuhan, China) provided the human renal proximal tubular epithelial cell line HK-2 (cat. no. CL-0109). The cells tested negative for the presence of mycoplasma and were authenticated by short tandem repeat profiling. Roswell Park Memorial Institute-1640 medium (11875119, Gibco, Rockville, MD, the USA) with 10% fetal bovine serum (A5670701, Gibco, Rockville, MD, the USA) was utilized for cell cultivation. Experimental conditions included maintenance at 37°C in a culturing incubator supplied with 5% carbon dioxide.
The AKI cell model was established by following a previously reported method with some improvements.[15] Different concentrations (0, 0.1, 1, 10, and 50 μg/mL) of lipopolysaccharide (LPS) (L5293, LPS; Sigma‒Aldrich, San Luis, MO, the USA) were used to induce HK2 cells for 24 h. In subsequent experiments, 10 μg/mL LPS was chosen to induce HK2 cells at different time points (0, 4, 8, 12, and 24 h) to select the optimal treatment duration.
Cell transfectionGeneral Biol (Chuzhou, China) synthesized two small interfering RNA (siRNA) fragments (ANXA3 siRNA1 and siRNA2) and the negative control (NC) siRNA. In accordance with the instructions included with the transfection reagent Lipofectamine 3000 (L3000001, Invitrogen, Karlsruhe, Germany), HK2 cells were cotransfected with ANXA3 siRNA1 (sense strand, 5'-GGAAUAUCAAGCAGCAUAU TT-3'), ANXA3 siRNA2 (sense strand, 5'-GGACAAG CAGGCAAATGAAGG-3'), or NC siRNA (sense strand, 5'-UUCUCCGAACGUGUCACGUTT-3') for 48 h. Functional experiments were conducted at 48 h after in vitro transfection.
Quantitative real-time polymerase chain reaction (qRT-PCR)Total RNA was isolated from cells using an RNA extraction kit (CW0591S, Cwbio, Beijing, China). Complementary DNA synthesis was then performed with Evo m-mlv reverse transcription reagent (AG11743, Accyrate Biology, Changsha, China). Subsequently, qRT-PCR was conducted on a qRT-PCR instrument (CFX96, Bio-Rad, Hercules, CA, the USA) by utilizing a SYBR Green Pro kit (AG11740, Accyrate Biology, Changsha, China). Primers included ANXA3 forward (5'- TCCGAAACATCTGGTGAC-3'), ANXA3 reverse (5'- TCAAGTTCTTCGTAAT ACCGAT-3'), GAPDH forward (5'-CACTACCGTACCTGACACCA-3'), and GAPDH reverse (5'- ATGTCGTTGTCCCACCACCT-3'). The 2−ΔΔCt method was utilized to calculate ANXA3 messenger ribonucleic acid (mRNA) expression. GAPDH was used as the internal control.
Measurement of cell proliferative potentialsThe cell counting kit (CCK)-8 assay was utilized to evaluate the viability of HK2 cells. HK2 cells (3 × 103 cells/mL) were seeded into 96-well plates and incubated for 0, 24, 48, 72, and 96 h before the addition of 15 μL of CCK-8 solution (CK04, Dojindo Laboratories, Shanghai, China). After a 2 h incubation period, cell viability was assessed with a microplate reader (cat. no. 1681130) from Bio-Rad Laboratories (Hercules, CA, the USA).
In addition, the growth potential of HK2 cells was evaluated by conducting colony formation assays. Approximately 1000 cells were seeded into the individual wells of six-well plates and cultured for 2 weeks, during which the growth medium was refreshed twice a week. The cell colonies that formed were subsequently treated with methanol, exposed to 0.1% crystal violet (548–62–9, Sigma‒Aldrich, St. Louis, Missouri, USA) for 20 min, and finally enumerated using a light microscope (Axio Scope A1; Carl Zeiss, Oberkochen, Germany) following washed.
A 5-ethynyl-2'-deoxyuridine (EdU) proliferation assay kit (cat. no. ab219801; Abcam, Cambridge, the UK) was used to evaluate EdU-positive HK2 cells. HK2 cells were exposed to 50 μM EdU and maintained at 37°C for 2 h. They were treated with a solution containing 4% formaldehyde (P0099, Beyotime, Shanghai, China) for fixation and permeabilized using 0.5% Triton X-100 (ST1722, Beyotime, Shanghai, China) for 20 min. Next, the cells were treated with a room-temperature Apollo reaction cocktail (×1; cat. no. C00003; RiBoBio, Guangzhou, China) for half an hour. Subsequently, DNA was stained with 4’,6-diamidino-2-phenylindole (cat. no. C1002; Beyotime, Shanghai, China) for an additional 30 min. Fluorescence microscopy (cat. no. Axio-Lab.A1-W-1; Carl Zeiss, Oberkochen, Germany) was employed to reveal the presence of EdU-positive HK2 cells (scale bar = 100 μm).
Measurement of inflammatory and oxidative stress indicatorsIn accordance with the guidelines provided by the manufacturer, specific enzyme-linked immunosorbent assay kits (Beyotime, Shanghai, China) were utilized to assess inflammatory responses in cells. Furthermore, the corresponding commercial assay kits from Beyotime were employed to measure the levels of superoxide dismutase (SOD) (cat. no. S0101S), reactive oxygen species (ROS) (cat.no. S0033S), glutathione (GSH) (cat. no. S0052), and malondialdehyde (MDA) (cat. no. S0131S) in HK2 cells.
Flow cytometryApoptotic HK2 cells were evaluated by utilizing an apoptosis detection kit (cat. no. BMS500FI-300) from Thermo Fisher Scientific (Rockford, MD, the USA). HK2 cells were suspended in PBS then stained with 10 μL of Annexin V-fluorescein isothiocyanate (FITC) (C1062S, Beyotime, Shanghai, China) and 5 μL of propidium iodide (ST1569, Beyotime, Shanghai, China) at 25°C for 15 min in the absence of light. Subsequently, an FACScan flow cytometer (cat. no. BDFACSAriaII; BD Biosciences, Franklin Lakes, NJ, the USA) was employed for the analysis of apoptotic cells.
Western blot analysisProteins were extracted from HK2 cells using radio-immunoprecipitation assay (RIPA) lysis buffer (AR0105, BOSTER, Wuhan, China). The proteins were initially separated through SDS polyacrylamide gel electrophoresis. Once the separation was complete, the proteins were employed for subsequent blotting with a PVDF membrane. The membrane was exposed to the primary antibodies anti-ANXA3 (cat. no. 11804-1-AP; Proteintech, Wuhan, China; dilution of 1:1000); anti-C/enhancer binding protein (EBP) homologous protein (CHOP) (cat. no. 15204-1-AP; Proteintech; dilution at 1:1000); anti-glucose-regulated protein 78 (GRP78) (cat. no. 11587-1-AP; Proteintech; dilution of 1:1000), anti-inositol-requiring enzyme 1α (IRE1α) (cat. no. 27528-1-AP; Proteintech; dilution of 1:1000), anti-activating transcription factor 6 (ATF6) (cat. no. 24169-1-AP; Proteintech; dilution of 1:1000), and anti-GAPDH (cat. no. Ab104788; Abclonal, Wuhan, China; dilution of 1:1000) at 4°C for 12 h after being blocked with nonfat milk (5%). Next, the membrane was incubated with the secondary antibody (cat. no. SA00001-1; Proteintech; dilution of 1:5000) for half an hour at 25°C. Immunoblotting was visualized using an ECL detection kit (cat. no. 180–501; Tanon, Shanghai, China).
Statistical analysisData analysis was conducted using the Statistical Package for the Social Sciences software v22.0 (IBM, Armonk, NY, the USA). Data were reported as the average value plus or minus the standard deviation. Disparities in the data were evaluated by utilizing one-way analysis of variance and Student’s t-tests, as deemed suitable. A P < 0.05 was deemed to be statistically significant.
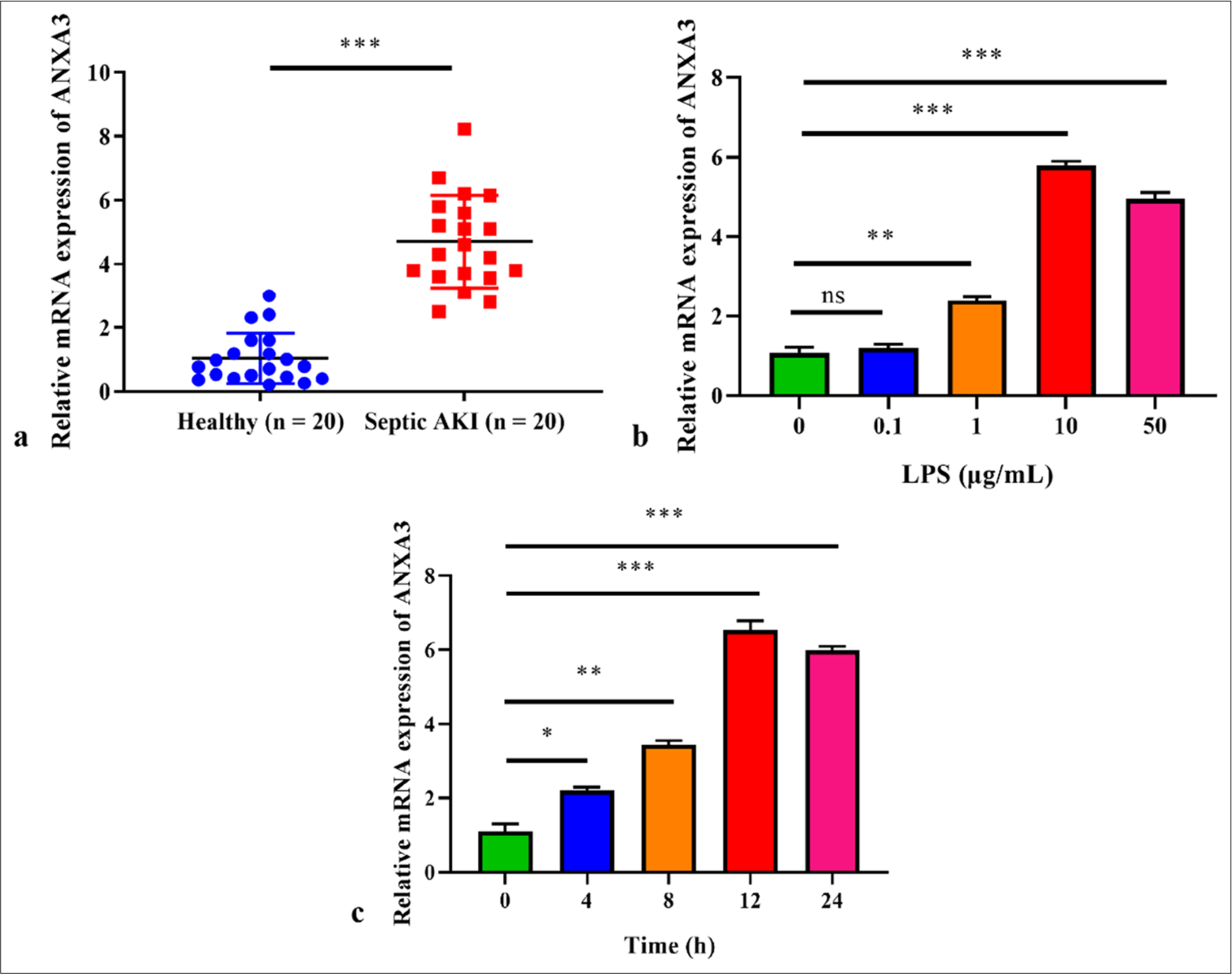
RESULTS High level of ANXA3 is observed in patients with sepsis and AKI and LPS-induced HK2 cellsCompared with healthy controls, patients with sepsis and AKI exhibited a notable elevation in serum ANXA3 levels [Figure 1a]. We also simulated AKI in vitro by exposing HK2 to LPS, and our findings supported that exposure to 1 μg/mL LPS resulted in a significant elevation in ANXA3 expression [Figure 1b]. Meanwhile, we noted an increase in ANXA3 expression that was dependent on the concentration of LPS following exposure (P < 0.001) and found no noticeable difference between the groups administered with 10 or 50 μg/mL LPS. Moreover, LPS (10 μg/mL) resulted in a time-dependent enhancement in ANXA3 expression, which reached its peak at 12 h post challenge [Figure 1c]. Therefore, the experiments described below involved the use of HK2 cells exposed to LPS (10 μg/mL) for 12 h.
Export to PPT
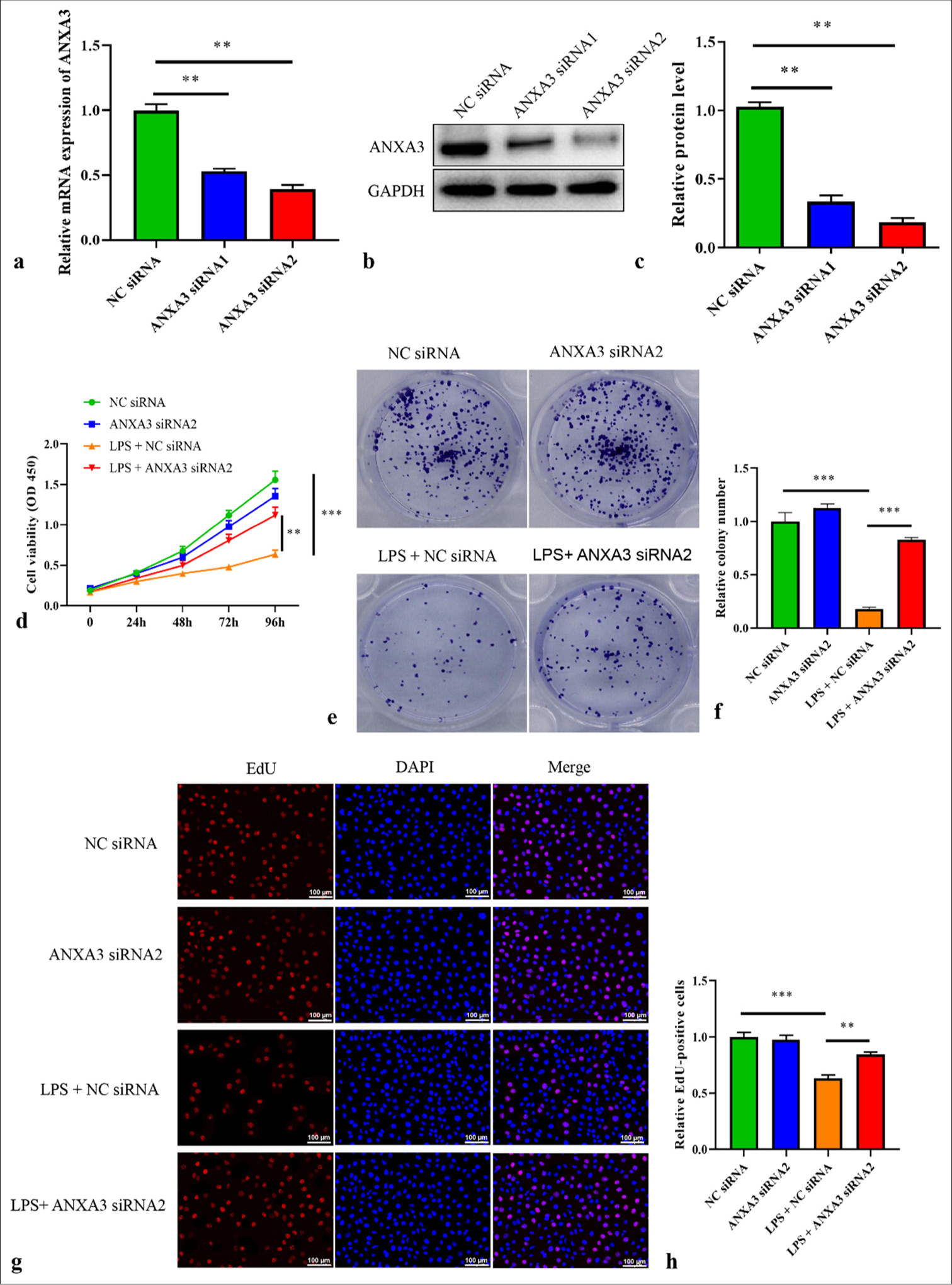
Knocking down ANXA3 induces the proliferative capacities of LPS-induced HK2 cellsANXA3 was knocked down using siRNA to investigate its function. Two siRNA fragments, namely ANXA3 siRNA1 and siRNA2, were used to reduce ANXA3 expression in HK2 cells effectively. Figure 2a shows that the reduction in ANXA3 mRNA expression was more pronounced in HK2 cells transfected with ANXA3 siRNA2 than in other cells, leading to the selection of these cells for subsequent experiments. Similar results were observed for the protein level of ANXA3 following transfection [Figure 2b and c]. The CCK-8 assay revealed a significant decrease in HK2 cell viability following LPS treatment [Figure 2d], whereas the transfection of ANXA3 siRNA2 remarkably restored the viability of LPS-induced HK2 cells. Similar patterns were observed in the colony numbers [Figure 2e and f] and EdU-positive cells [Figure 2g and h] of different groups. These results suggested that LPS-induced HK2 cells transfected with ANXA3 siRNA2 had enhanced proliferative capacities.
Export to PPT
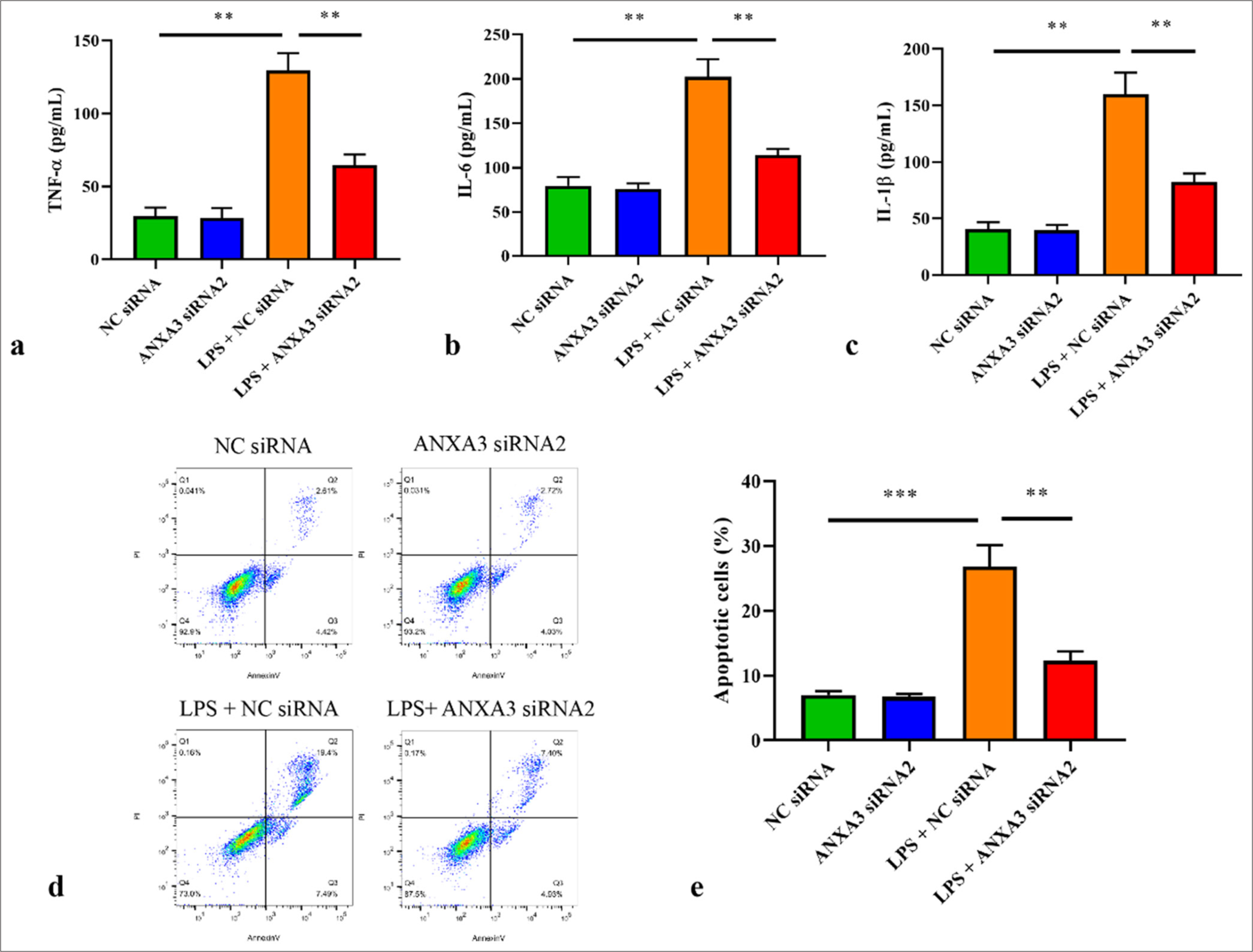
Silencing of ANXA3 suppresses inflammatory responses and apoptosis in vitroAs indicated in Figure 3a-c, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and IL-1β amounts significantly increased in HK2 cells following LPS exposure but were largely inhibited by ANXA3 silencing. In addition, the apoptosis of HK2 cells stimulated by LPS was assessed through flow cytometry. We demonstrated that LPS treatment significantly induced HK2 cell apoptosis, whereas ANXA3 knockdown remarkably inhibited LPS-induced apoptosis in HK2 cells [Figure 3d and e].
Export to PPT
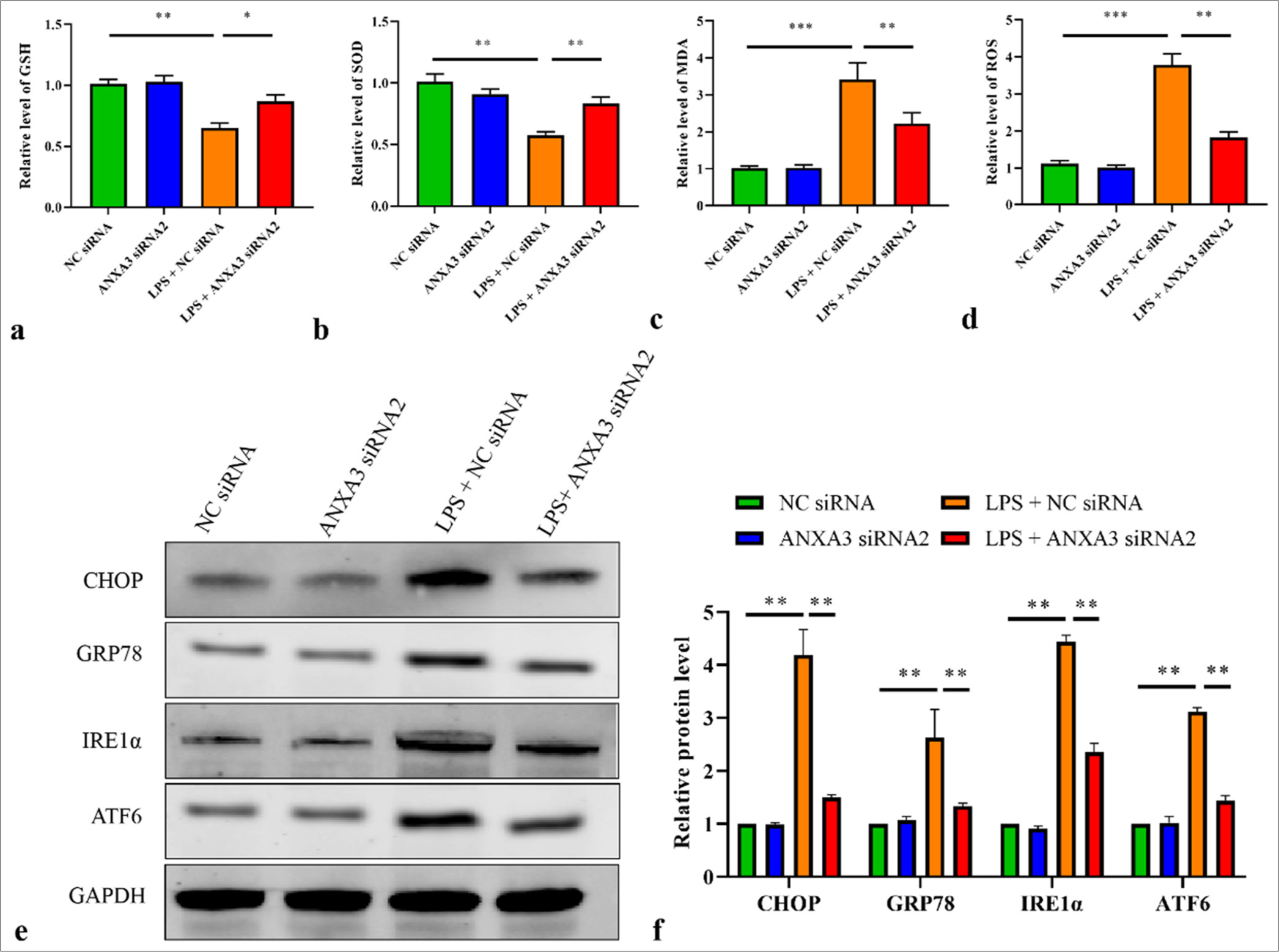
Knockdown of ANXA3 attenuates oxidative and ER stresses in vitroThe next step included assessing the effects of ANXA3 on oxidative and ER stresses. GSH and SOD amounts in LPS-induced HK2 cells were significantly repressed but markedly recovered following the transfection of ANXA3 siRNA2 [Figure 4a and b]. Conversely, MDA and ROS activities in HK2 cells were dramatically increased by LPS treatment, whereas MDA and ROS amounts were notably suppressed by ANXA3 silencing [Figure 4c and d]. Furthermore, ER stress markers, such as ATF6, IRE1α, GRP78, and CHOP, were quantified by using Western blot analysis. Figure 4e and f illustrate that LPS treatment could enhance the protein levels of ATF6, IRE1α, GRP78, and CHOP in HK2 cells, whereas knocking down ANXA3 significantly inhibited their expression.
Export to PPT
DISCUSSIONOn the basis of current estimates, it is projected that approximately 19 million sepsis cases are expected to occur globally annually, resulting in up to 11 million patients developing sepsis-induced AKI each year.[16] Furthermore, when compared with AKI caused by other factors in critically ill individuals, sepsis-induced AKI is linked to extended hospitalization, elevated treatment expenses, and heightened likelihood of death.[16] Despite the development of various supportive measures, such as plasma perfusion, renal replacement therapy, fluid balance management, and hemodynamic stability maintenance, for sepsis-induced AKI, the overall mortality rate of this syndrome remains high.[4] In this study, we preliminarily elucidated a novel mechanism attenuating the development of sepsis-induced AKI, indicating that the knockdown of ANXA3 can ameliorate apoptosis, inflammation, ER stress, and oxidative stress in sepsis-induced AKI in vitro.
LPS is well established to be associated with renal tubular epithelial cell apoptosis.[17] Research has shown that LPS is commonly used to mimic AKI in animal and cell culture models.[18] In this study, an LPS-induced HK2 cell model was established and a significant increase in the expression of ANXA3 was observed. Moreover, serum ANXA3 level was higher in patients with sepsis and AKI than in healthy controls. As reported by published data, high ANXA3 level has been identified as a diagnostic biomarker of pediatric patients with sepsis.[13] Furthermore, it is closely linked to the emergence of sepsis-related acute lung damage, which serves as another deadly complication of sepsis.[14] We therefore speculated that high ANXA3 levels may contribute to the development of sepsis-induced AKI as a potential pathogenic factor. A growing number of investigations have posited that the development of AKI is closely linked to epithelial cell apoptosis, inflammation, and ER and oxidative stresses.[19,20] In the progression of sepsis-induced AKI, an overabundance of inflammation triggers ROS generation. These ROS target polyunsaturated fatty acids in biofilms, resulting in a notable increase in MDA production. MDA amounts serve as an indicator of the extent of ROS damage to cells.[21] SOD and GSH play vital parts in scavenging oxygen free radicals from cells. Herein, our findings demonstrated that LPS treatment remarkably promoted the secretion of TNF-α, IL-6, and IL-1β. Moreover, after HK-2 cells were exposed to LPS, their levels of ROS and MDA increased, whereas their contents of SOD and GSH reduced. These obtained data evidenced an imbalance between endogenous oxidases and nonenzymatic antioxidants, leading to the onset of oxidative stress. However, ANXA3 silencing markedly reversed the above changes, indicating its anti-inflammation and antioxidant effects in sepsis-induced AKI in vitro.
The development of sepsis-induced AKI is complicated. The occurrence of ER stress and overproduction of inflammatory factors can also lead to renal cell apoptosis. In pathological circumstances, multiple factors that promote stimulation could lead to the buildup of improperly folded proteins in the ER. This phenomenon can cause dysfunction and trigger stress within the ER. An overabundance of ER stress has the potential to induce the expression of CHOP, a gene that promotes apoptosis, ultimately leading to cellular apoptosis.[20] In this study, we found that LPS challenge not only stimulated apoptosis in HK2 cells but also elevated the amounts of ER stress proteins (ATF6, IRE1α, GRP78, and CHOP). As anticipated, the knockdown of ANXA3 attenuated the promoting effects of LPS treatment on ER stress and apoptosis in HK2 cells. Overall, our findings suggested that ANXA3 silencing alleviates sepsis-induced AKI by suppressing apoptosis, inflammation, ER stress, and oxidative stress in vitro.
Some limitations of this study should not be ignored. First, at least two established cell lines must be used to perform experiments. Second, exploring the effects of ANXA3 overexpression on sepsis-induced AKI in vitro may improve the rigor of this study. Third, the findings of this study should be validated by experiments on animal models.
SUMMARYANXA3 levels in LPS-induced HK2 cells and serum samples obtained from patients with AKI and sepsis considerably increased. Interference with ANXA3 remarkably mitigated LPS-induced HK2 cell injuries, including inflammation, oxidative stress, apoptosis, and ER stress. The findings of this work may provide valuable insights for the development of clinical diagnosis and therapeutic strategies for sepsis-induced AKI.
Comments (0)