
Remember me
In the past decades, lung cancer has been the most frequently diagnosed cancer and the primary cause of cancer-related mortality globally.[1] It is initially asymptomatic and typically discovered at advanced stages[2] and is often associated with risk factors, such as long-term smoking; exposure to second-hand smoke, environmental toxins, and ionizing radiation; human immunodeficiency virus infection; alcohol consumption; and genetic factors.[3] Lung cancer treatments include surgical removal, chemotherapy, radiation therapy, and therapy targeting specific cancer characteristics.[4] Treatment selection is influenced by patients’ conditions, tumor type and stage, and overall health.[5] However, the 5-year survival rate of lung cancer remains poor.[6] Metastasis and resistance to treatment present obstacles to lung cancer management.[7] Thus, elucidating the pathogenesis and progression of lung cancer is imperative.
Tumor-associated endothelial cells (TAECs) are crucial for tumor development and metastasis, possessing the following unique characteristics: (1) Enhanced angiogenic capacity: Stimulated by hypoxia and inflammation within the tumor microenvironment, TAECs overexpress angiogenic factors, and promoting neovascularization. TAECs enhance this capability through autocrine and paracrine mechanisms.[8] (2) Impaired barrier function: The reduced expression or functionality of junction proteins in TAECs diminishes vascular integrity and permeability.[9] (3) Endothelial–mesenchymal transition (EndMT) characteristics: TAECs can undergo EndMT, gaining migratory and invasive capabilities, facilitating tumor vascular remodeling and metastasis, which is characterized by a decrease in endothelial cell markers and an increase in mesenchymal cell markers.[10] These traits collectively favor tumor growth and metastasis.[11]
Exosomes (30–150 nm) are small extracellular vesicles released by various cells, including normal and cancer cells, into the extracellular environment.[12] They are among the components of the body’s intercellular communication system and deliver multiple functional biomolecules, including proteins, lipids, and nucleic acids.[13] Most cells release exosomes, whereas tumor cells may produce more exosomes to promote their own survival and invasion under hypoxic conditions.[14] Some researchers have highlighted the role of hypoxic tumor cell-derived exosomes in promoting angiogenesis.[15-17]
A microRNA (miRNA) operates by specifically attaching to a messenger RNA (mRNA), resulting in the degradation of the mRNA or suppression of its translation into a protein.[18] Cancer-derived exosomal miRNAs are involved in cancer metastasis and progression. For example, exosomal microRNA-5703 (miR-5703) has been demonstrated as an onco-miRNA in pancreatic cancer.[19] However, the function of exosomal miRNAs has not been fully characterized in lung cancer angiogenesis, tumor growth, and metastasis.
In this study, we investigated the expression pattern of miR-5703 in lung cancer and determine whether lung cancer cell-derived exosomes can induce the angiogenesis, tumorigenesis, and EndMT of TAECs. We showed that exosomal miR-5703 down-regulated the expression of inhibitor of growth family member 4 (ING4) in endothelial cells and thereby promoted lung cancer progression and angiogenesis. These results strongly indicate that exosomal miR-5703 is a prognostic indicator for tumor progression and a therapeutic target.
MATERIAL AND METHODS Sampling from patientsA cohort of patients with lung cancer was meticulously selected using stringent criteria. Each patient underwent an exhaustive pre-operative clinical evaluation encompassing chest radiography, computed tomography, and pulmonary function test for the accurate diagnosis of lung carcinoma and precise delineation of tumor dimensions and locations. Apart from diagnostic modalities, an in-depth collection of patients’ clinical attributes was undertaken. The data encompassed age, gender, metastatic status, tumor dimension, and clinical stage. On the confirmation of lung cancer diagnosis, patients were apprised in detail regarding the study’s objectives and methodologies. The study was conducted in compliance with the Declaration of Helsinki. Informed consent was obtained from all study participants, and ethics approval was obtained from the Ethics Committee of the Second People’s Hospital of Yibin (no. 2023-162-01). During surgical intervention, paired tissue specimens, cancer tissues (n = 20), and adjacent normal tissues (n = 20) were harvested. Blood samples were drawn from each patient or healthy individual (n = 20), and serum samples were isolated through centrifugation. Serum exosomes were isolated using an exosome isolation kit (4478360, Invitrogen, Massachusetts, USA). As for the tissues, potential RNA degradation was prevented by immediately immersing the specimens in an RNA preservation solution (R0118, Beyotime, Shanghai, China). Serum blood samples were harvested, aliquoted, and stored at −80°C. Patient data for the tissue samples are shown in Table 1.
Table 1: Patient data for the tissue samples.
No Age Sex Diagnosis Metastasis Tumor size (diameter, cm) Stage 1 61 M Squamous cell carcinoma Absent ≤3 IB 2 59 M Squamous cell carcinoma Absent >3 IIA 3 67 F Bronchioloalveolar carcinoma Absent ≤3 IA 4 47 F Adenosquamous carcinoma Present >3 IIIB 5 64 M Adenocarcinoma Absent >3 IIB 6 64 M Squamous cell carcinoma Absent >3 IIA 7 51 F Small-cell carcinoma Present >3 IVB 8 48 M Squamous cell carcinoma Absent ≤3 IB 9 62 M Adenocarcinoma Absent >3 IIA 10 63 F Mucoepidermoid carcinoma Present >3 IIIA 11 59 M Mucoepidermoid carcinoma Absent ≤3 IB 12 71 M Squamous cell carcinoma Present >3 IIIA 13 52 M Squamous cell carcinoma Absent >3 IIA 14 66 F Adenocarcinoma Absent ≤3 IA 15 65 F Small-cell carcinoma Absent ≤3 IB 16 41 M Adenocarcinoma Absent ≤3 IB 17 53 M Mucoepidermoid carcinoma Absent >3 IIA 18 43 M Mucoepidermoid carcinoma Absent ≤3 IB 19 48 M Bronchioloalveolar carcinoma Present >3 IIB 20 59 M Bronchioloalveolar carcinoma Present >3 IIB Cell culture and co-cultureHuman bronchial epithelial cell line (Bronchial Epithelial Cell Line 2B [BEAS2B], Delf-10130) and 95D cell line (Delf-10140) were newly purchased from the Hefei Wanwu Biotechnology Co., LTD; non-small-cell lung cancer cell lines A549 (1101HUM-PUMC000002), (Human lung cancer cell line 827, 1101HUM-PUMC000478), (National Cancer Institute-H1299, 1101HUM-PUMC000469), and (National Cancer Institute-H1650, 1101HUM-PUMC000251); lung squamous carcinoma cell line (Squamous Cell Carcinoma-MES 1, 1101HUM-PUMC000262); and small-cell lung cancer cell line (National Cancer Institute-H1688, 3101HUMTCHu154) were purchased from the National Infrastructure of Cell Line Resource of China (Beijing, China). A certificate of analysis confirming cell line identity through short tandem repeat profiling and confirming lack of mycoplasma contamination was obtained. The cells were cultured under 37°C in an environment with saturated humidity and 5% Carbon dioxide (CO2) in a Roswell Park Memorial Institute (RPMI) 1640 medium (C0893, Beyotime, Shanghai, China) enriched with 10% Fetal Bovine Serum (FBS; C0252, Beyotime, Shanghai, China) and 100 U/mL penicillin–streptomycin (C0222, Beyotime, Shanghai, China). Appropriate nutrition and pH balance were ensured by replacing the medium every 2–3 days.
Interactions between lung cancer and endothelial cells were evaluated with a Transwell chamber co-culture system (3422, Corning, New York, USA). Lung cancer cells were seeded in the bottom chamber, and TAECs were positioned in the top chamber in a ratio of 5:1. Afterward, the chambers were incubated under standard culture conditions: temperature of 37°C, 100% humidity, and atmosphere containing 5% CO2.
TAEC isolationTAECs were isolated from resected lung carcinoma specimens obtained immediately after surgical removal.[20] The specimens were meticulously minced and enzymatically dissociated in an RPMI 1640 medium enriched with 0.1% collagenase IV (C5138, Sigma–Aldrich, Missouri, USA) at 37°C for 1 h. The resultant cellular suspension was sequentially filtered through a series of graded meshes, and stromal and aggregated materials were discarded. TAECs were enriched using anti-CD34 monoclonal antibodies (ab81289, Abcam, Massachusetts, USA) bound to magnetic beads, and isolation was carried out with a magnetic cell sorting (MACS) system (Miltenyi Biotech, Bergisch Gladbach, Germany). Diphtheria toxin (500 ng/mL, Calbiochem, San Diego, CA, USA) was added to the TAEC cultures to eliminate remaining human tumor cells. After 24 h, dead cells were aspirated, and non-endothelial tumor cells were completely removed. The TAECs were further purified using a second MACS step, and Fluorescein Isothiocyanate-labeled BS1-B4 Lectin (FITC-BS1-B4 lectin) was used to eliminate contaminating stromal cells and enhance the purity of the endothelial cells. The purity of the isolated TAECs was confirmed through flow cytometric (FCM) analysis. Markers, such as FITC-BS1-B4, were used to ensure that the cell population was primarily composed of endothelial cells. After isolation, the TAECs were tested and found negative for mycoplasma contamination. The TAECs were grown in endothelial growth medium-2 (CC-3162, Lonza Group Ltd., Basel, Switzerland), which was enhanced with 10% FBS and 100 U/mL penicillin–streptomycin. The cultured TAECs were then passaged at subconfluency, and experiments were conducted using cells from passages 1–6 to ensure cellular fidelity.
Exosome extraction and characterization detectionAfter tumor cells were transfected with miR-5703 mimics, inhibitors, and negative control (NC) (GenePharma, Suzhou, China), the culture supernatant was collected and retained after 10 min of centrifugation at ×350 g. Then, the supernatant was centrifuged again (×2000 g for 30 min). The supernatant was mixed with an exosome extraction working solution (C10130-1, Ribo Bio, Guangzhou, China) and left overnight at 4°C. The next day, the samples were centrifuged, and the lower sediment was retained. The exosomes were reconstituted in an optimal volume of Phosphate-Buffered Saline (PBS) and transferred to a cuvette. Exosome particle size analysis was carried out using a nanoparticle size analyzer (N30E, Xiamen Fuliu Biological Technology Co., Ltd, Xiamen, China). Exosome markers (CD81, CD63, and Alix) and negative markers (calnexin and α-tubulin) were detected by Western blotting. In addition, CD81 (ab79559, Abcam, Massachusetts, USA) was confirmed by FCM.[21] The method for isolating exosomes from patient sera was the same as that used for isolating exosomes from cell supernatants.
Exosomes were isolated and then fixed using 100 μL of 2.5% glutaraldehyde (G1102, Servicebio, Wuhan, China) and kept at 4°C. An exosome suspension (5 μL) was gently placed on a copper grid, where the exosomes were allowed to adhere for 20 min at room temperature. Subsequently, the grids were rinsed with PBS and then fixed with 1% glutaraldehyde for 5 min. The samples were rinsed 10 times with double- distilled water and negatively stained with 4% uranyl acetate (Hubei Xingyinhe Chemical Co., Ltd. Wuhan, China) for 5 min. Any excess staining solution was carefully dabbed off with filter paper, and the samples were allowed to dry at ambient temperature. The exosomes were visualized with a transmission electron microscope (H-7000; Hitachi, Ltd., Tokyo, Japan). The reagents, including the culture medium, FBS, and liquid used in this experiment, were exosome free.
Transwell assayTAECs were seeded into six-well plates at a density of 2 × 105 cells per well and cultured until they reached roughly 70% confluency. The isolated exosomes were then added to the TAECs at a concentration of 50 μg/mL, and the culture was maintained for 24–48 h at 37°C in a 5% CO2 atmosphere. Subsequently, a cell suspension consisting of either endothelial cells or lung cancer cells was prepared at a concentration of 1 × 106 cells/mL. This cell suspension was then added to the Transwell migration chamber (3428, Corning, New York, USA), typically dispensing 500 μL per well. The lower chamber was either seeded with an alternate cell type (TAECs or lung cancer cells) or left cell free. The Transwell chamber was incubated at 37°C with 100% humidity. After 48 h of incubation, the upper chamber was collected. Cells that did not migrate from the bottom of the inner chamber were scraped off, retaining only the cells that had migrated through the membrane. The cells were visualized through underwent 10 min 2% crystal violet staining, which were subsequently counted and imaged under a microscope. The migration behavior of the cells was then analyzed. To assess the invasive capability of lung cancer cells, we added a layer of Matrigel matrix (C0372, Beyotime, Shanghai, China) to the bottom of the Transwell chamber. On the basis of the number of cells that penetrated this matrix, the invasive potential of the cells was evaluated.
Cell proliferation assayCell viability was determined using a cell counting kit 8 (CCK8) kit (CCK-8, Dojindo, Kumamoto, Japan) and 5-ethynyl-2'-deoxyuridine (EdU) staining. After the cells were transferred with miRNA mimics, inhibitors, or plasmids, the cells were prepared for cell viability detection with the CCK8 kit or EdU staining.
CCK8Cell viability was assessed through a CCK-8 assay. Log-phase cells were harvested and resuspended to achieve a density of 10,000 cells/well in a 96-well plate. Each well was filled with 100 μL of the cell suspension. The plate was then incubated at 37°C in a 5% CO2 atmosphere for 24 h. After incubation, 10 μL of a CCK-8 solution was introduced to each well, and the plate was further incubated for 1 h. Finally, the absorbance of each well was measured at 450 nm with a microplate reader (Thermo Fisher Scientific, mμltiscan MK3).
EdU stainingCell proliferation was assessed using an EdU staining kit (C0075L, Beyotime, Shanghai, China). The cells were treated with 50 μM EdU for 2 h at 37°C, fixed with 4% paraformaldehyde (G1101, Servicebio, Wuhan, China), and permeabilized with 0.3% Triton X-100 (GC204003, Servicebio, Wuhan, China). Subsequently, the cells were exposed to a click reaction mixture for 30 min in accordance with the manufacturer’s guidelines. Finally, the cells were stained with 4',6-diamidino-2'-phenylindole (DAPI, C1002, Beyotime, Shanghai, China), and the proportion of EdU- positive cells, which is indicative of proliferating cells, was assessed through fluorescence microscopy (Eclipse 80i, Nikon, Tokyo, Japan).
Cell apoptosis detectionHoechst: Initially, cover slips were sterilized in 70% ethanol for at least 5 min and then air-dried or rinsed 3 times with PBS. After washing with the cell culture medium, cover slips were placed in six-well plates, and the cells were seeded and grown to 50–80% confluency. To measure cell apoptosis, we replaced the medium with 0.5 mL of fixative. After fixation, the cover slips were washed twice with PBS for 3 min each. The cells were then stained with 0.5 mL of Hoechst 33258 dye (C1011, Beyotime, Shanghai, China) for 5 min, agitated, and washed twice. Subsequently, the cells were stained with 0.5 mL of DAPI staining solution (C1002, Beyotime, Shanghai, China) for 5 min and washed twice. An antifade mounting medium was applied for microscopy. The stained cell nuclei appeared blue under a fluorescence microscope at excitation and emission wavelengths of approximately 350 and 460 nm, respectively.
Flow Cytometry (FCM)Cells were digested, centrifuged at ×500 g for 5 min at 4°C, and washed twice with PBS. Afterward, the cells were resuspended in ×1 binding buffer to a final volume of 5 × 106/mL, and 100 μL of the suspension was treated with 5 μL of annexin V–fluorescein isothiocyanate (C1062S, Beyotime, Shanghai, China) and 5 μL of propidium iodide (PI, C1062S, Beyotime, Shanghai, China). The suspension was then incubated in the dark at room temperature for 8–10 min. After 400 μL of ×1 binding buffer was added, the mixture was analyzed through FCM within an hour. The apoptosis rate was determined by the sum percentages of annexin V+/PI+ and annexin V+/PI- cells.
Immunofluorescence assayAfter co-cultivation with lung cancer cells, the expression of α-smooth muscle actin (α-SMA) and ING4 in endothelial cells was evaluated through immunofluorescence. Fixed with 4% paraformaldehyde for 15 min. Then, the cells were infiltrated with 0.1% Triton X-100 for 5 min and blocked with 1% BSA at room temperature for 1 h. Primary antibodies targeting ING4 (1:200, ab113425, Abcam, Massachusetts, USA) and (α-SMA, 1:100, ab7817, Abcam, Massachusetts, USA) were applied to the cells, which were incubated overnight. Then, the cells were incubated with suitable secondary antibodies (1:1000, ab150077, Abcam, Massachusetts, USA). Finally, the cells were stained with DAPI (C1002, Beyotime, Shanghai, China) and examined using a confocal laser scanning microscope (OLS5000, Olympus, Tokyo, Japan). The expression levels of ING4 and α-SMA in the endothelial cells were examined using images.
Tube formation assayEndothelial cells, on reaching 80% confluency, were washed with PBS once or twice and then treated with trypsin (P4201, Beyotime, Shanghai, China). They were then incubated at 37°C for 2–3 min. To neutralize trypsin, a fresh complete medium was introduced, and the cells were resuspended to yield a single-cell suspension. The cell density was adjusted to 1 × 106 cells/mL. Subsequently, 100 μL of the cell suspension with 1 × 105 cells/mL was plated into a six-well plate and cultured overnight at 37°C in a 5% CO2 atmosphere. The next day, after specific treatments, the cells were incubated for 48 h. Concurrently, Matrigel was thawed overnight at 4°C. All materials, including pipettes and tips, were prechilled. With a cold pipette tip, 100 μL of Matrigel was added to a prechilled 48-well plate, and even distribution in the wells was ensured. After the application of Matrigel to the plate, the plate was incubated for 40 min at 37°C for the solidification of the Matrigel matrix. Subsequently, the cells were counted, and their density was adjusted to 3 × 105 cells/mL. The Matrigel-coated 48-well plate was washed with PBS 3 times. The volume of 200 μL of the cell suspension was 6 × 104 cells. The suspension was dispensed into each well until the density was 8 × 104 cells per well. The plate was then maintained at 37°C in an atmosphere containing 5% CO2. After 5 h, tube formation was inspected using an inverted microscope, and images were obtained for subsequent analysis.
Quantitative real-time polymerase chain reaction (qRT-PCR)Total RNA was isolated from the collected clinical tissues and cell lines with TRIzol reagent (15596026, ThermoFisher, Illinois, USA). A reverse transcription kit (G3335-50, Servicebio, Wuhan, China) was used in synthesizing complementary DNA (cDNA). For qRT-PCR detection, a reaction mixture consisting of cDNA, SYBR Green fluorescent dye and specific primers was prepared using SYBR Green I Master Mix on a 7900 sequence detector (Applied Biosystems, Foster City, CA, USA). The conditions for qRT- PCR were as follows: Initial denaturation step at 95°C for 10 min for 40 cycles. Each cycle consisted of a denaturation phase at 95°C for 15 s and an annealing/extension phase at 60°C for 1 min. The data were analyzed using the 2−ΔΔCT method.[22] For miRNAs, U6 served as the internal reference. As for mRNAs, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was utilized as the internal reference. The primer sequences in RT-PCR are provided in Table S1.
Western blotProtein lysates from tissues or cells were prepared using a radio-immunoprecipitation assay buffer (G2002, Servicebio, Wuhan, China) containing protease and phosphatase inhibitors (G2006, Servicebio, Wuhan, China). Protein concentrations were measured with a bicinchoninic acid assay. Equal amounts of the proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membranes (G6015, Servicebio, Wuhan, China). These were incubated overnight at 4°C with primary antibodies against E-cadherin (1:1000; ab227639; Abcam), N-cadherin (1:1000; ab76011; Abcam), vimentin (1:1000; EPR3776; Abcam), Matrix Metalloproteinase-2 (MMP2, 1:1000; ab92536; Abcam), Matrix Metalloproteinase-9 (MMP9, 1:1000; ab76003; Abcam), and GAPDH (1:1000; ab8245; Abcam). After incubation with primary antibodies, the membranes were washed and treated with Horseradish Peroxidase -linked secondary antibodies (1:5000, ab205718, Abcam, Massachusetts, USA), and bands were detected using an enhanced chemiluminescence system (G2020, Servicebio, Wuhan, China). Band intensities were quantified, and expression was normalized to that of GAPDH.
In vivo modelBreeders’ Association of Laboratory Animals, California nude mice (6–8 weeks old, approximately 20 g, n = 20) were purchased from Animal Center of Tianjin Medical University. They were subcutaneously injected with 5 × 106 H1299 cells each. The mice were divided into four groups (n = 5) and received intratumoral injections every 3 days: 100 μL PBS (the blank group), H1299-derived exosomes (the control group), exosomes with miR-5703 inhibitor NC (the NC group), and exosomes with miR-5703 inhibitor (the inhibitors group). Tumor volumes were measured weekly with the formula V = width2 × length/2 (mm3). On day 28, mice were euthanized after 1% pentobarbital sodium anesthesia (50 mg/kg). Tumors were collected, weighed, and prepared for histological analysis. For hematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and immunohistochemistry, the tissues were fixed in 4% paraformaldehyde. For quantitative PCR (qPCR) and Western blot, the samples were stored at −80°C. Ethics approval was obtained from the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (approval No. 2023078). All animal experiments were conducted in strict accordance with the guidelines for the care and use of laboratory animals established the Second People’s Hospital of Yibin. All procedures were performed under appropriate anesthesia, and all animals received humane care throughout the duration of the study.
H&E stainingThe tissues were fixed in paraformaldehyde, embedded in paraffin, and sectioned continuously (4 μm). After dehydration and dewaxing, the sections were sequentially stained with H&E (G1076, Servicebio, Wuhan, China), mounted with neutral resin, and examined under a bright- field microscope (Eclipse 80i, Nikon, Tokyo, Japan) for histopathological analysis.
ImmunohistochemistryAfter dehydration and dewaxing, the tissue sections were subjected to antigen retrieval and peroxidase blocking. After blocking with serum, the sections were incubated overnight with the following primary antibodies: E-cadherin (1: 100, ab227639; Abcam), N-cadherin (1: 50, ab76011, Abcam), Ki67 (1:500, ab15580; Abcam), and CD34 (1:200, ab81289; Abcam). Then, the sections were incubated with secondary antibodies for 1 h (1:5000, ab205718, Abcam). After developing with 3,3’-Diaminobenzidine (G1212, Servicebio, Wuhan, China), the sections were counterstained with hematoxylin, mounted with neutral resin, and examined under a bright-field microscope (Eclipse 80i, Nikon, Tokyo, Japan) for the assessment of protein expression levels and localization.
TUNEL assayAfter dehydration and dewaxing, the tissue sections were treated with proteinase K (MK1012-100, Boster Biotechnology, Wuhan, China). After thorough washing with PBS, a TUNEL assay reagent consisting of Terminal Deoxynucleotidyl Transferase enzyme, fluorescence labeling solution, and TUNEL detection solution (MK1012-100, Boster Biotechnology, Wuhan, China) was applied. The sections were then incubated at 37°C for 60 min, extensively washed with PBS, and finally mounted using an antifade mounting medium to prevent fluorescence quenching. The sections were then observed with a fluorescence microscope (Eclipse 80i, Nikon, Tokyo, Japan). The excitation and emission wavelengths of Cy3 were 550 and 570 nm, respectively, resulting in red fluorescence.
Luciferase assayFor ING4 and has-miR-5703 luciferase assay, the cells were co-transfected with 50 nM miR-5703 mimics and 200 ng of a pmirGLO-ING4 vector or corresponding mutant type. A dual luciferase assay kit (Promega, WI, USA) was used for dual luciferase assay. At 48 h post-transfection, cells were collected, and Renilla luciferase activity was assessed. Results were assessed as the ratios of Firefly luciferase activity to Renilla luciferase activity.
Data collection and bioinformatics analysisTo explore the potential mechanisms of circulating exosomal biomarkers in the plasma of patients with lung cancer, we selected the GEO Series 198959 (GSE198959) dataset to analyze the differentially expressed miRNAs in the Exos of patients with recurrent lung cancer. At an adjusted P < 0.05 and |log2 (Fold Change)| of >1, highly expressed miRNAs were screened for subsequent validation. Kaplan–Meier curves were generated using Kaplan–Meier Plotter (KMplot, www.kmplot.com).[23] The gene expression data and relapse- free and Overall Survival information in the KMplot program were downloaded from Gene Expression Omnibus (GEO), European Genome-phenome Archive, and The Cancer Genome Atlas.
Data analysisData were presented as mean ± Standard deviation. The Statistical Package for the Social Sciences 19.0 (IBM, Armonk, NY, USA) was used for statistical analysis. Analysis of variance and Turkey’s post hoc tests were used for multiple- group comparisons, and t-test was utilized for two-group comparisons. P < 0.05 indicated statistical significance. Image editing was conducted using GraphPad Prism 9.0 (GraphPad Software Inc., La Jolla, CA, USA).
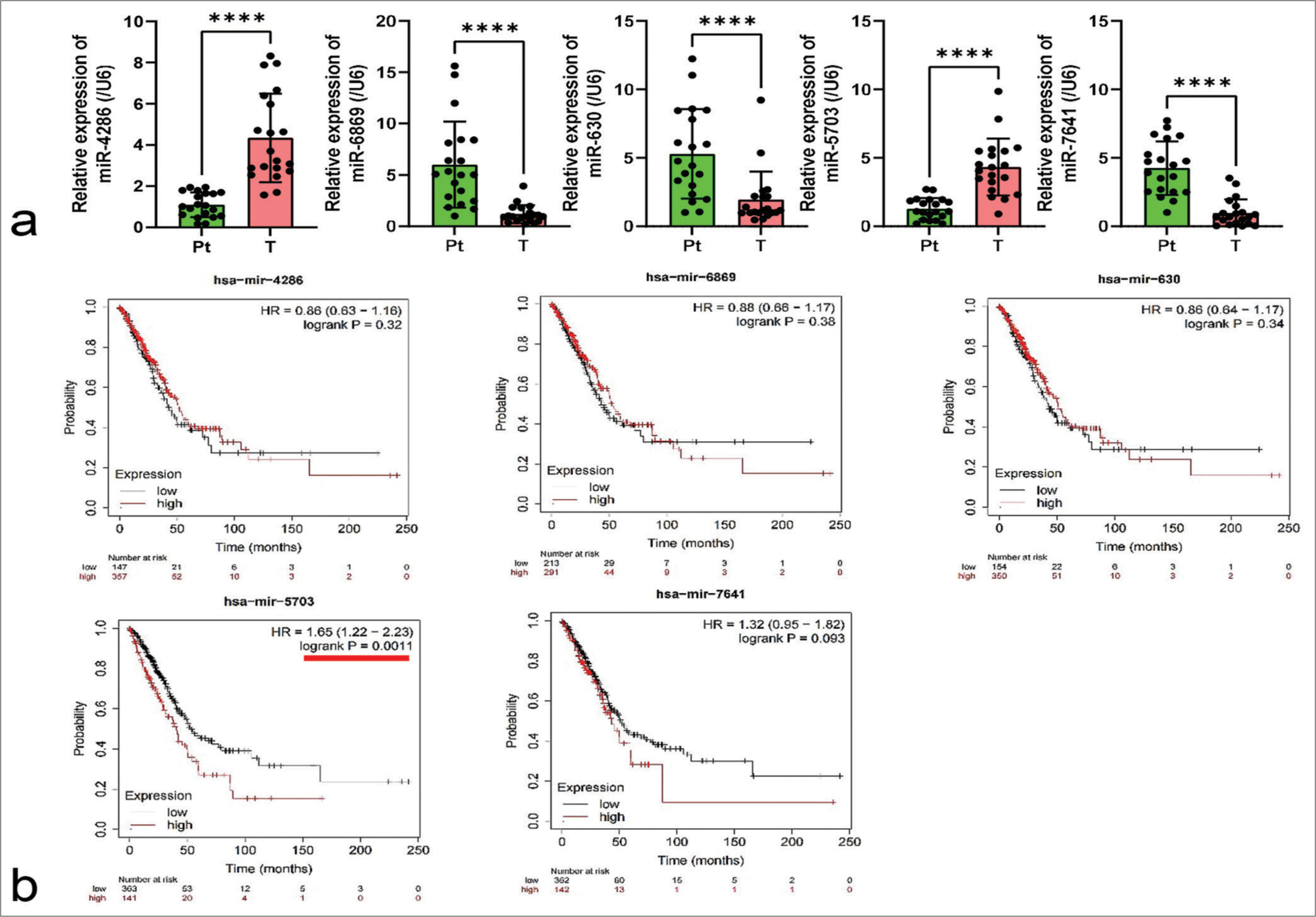
RESULTS Expression characteristics of miR-5703 in lung cancerIn a bid to identify potential factors in lung cancer, a study was conducted based on the GEO dataset database, using the GSE198959 dataset. Five miRNAs, namely, miR-4286, miR-6869, miR-630, miR-5703, and miR- 7641, were identified. By performing qPCR detection on clinical lung cancer tissues (T) and peri-cancerous tissues, it was observed that the expression levels of the miRNAs significantly varied in the lung cancer tissues (P < 0.0001), [Figure 1a].
Export to PPT
Further, employing the Kaplan–Meier Plotter platform (http://kmplot.com/analysis/index.php?p=background), the correlation of miR-4286, miR-6869, miR-630, miR-5703, and miR-7641 with lung cancer patients was respectively analyzed. Results revealed a significant correlation between miR-5703 and patient prognosis (P = 0.0011), [Figure 1b].
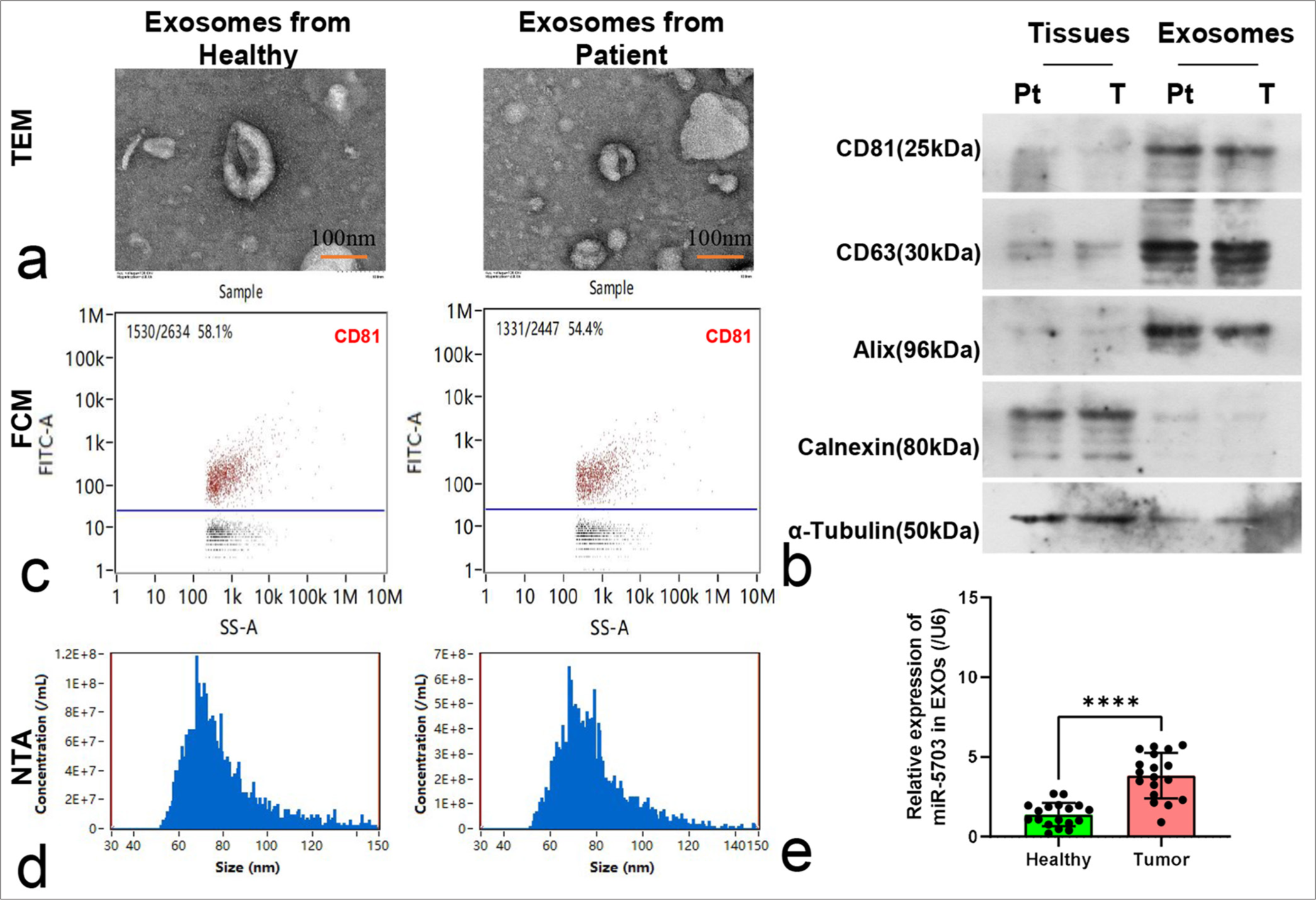
Expression characteristics of miR-5703 in exosomes of patients with lung cancerExosomes were isolated from the serum samples of healthy individuals and patients with lung cancer for characterization and detection. Electron microscopy observation showed that the exosomes were approximately 100 nm in size [Figure 2a]. Western blotting revealed the presence of CD81, CD63, and Alix and the absence of calnexin and α-tubulin within the exosomes [Figure 2b]. FCM tests showed a clear CD81-positive marker in the exosomes [Figure 2c].
Export to PPT
Subsequent analysis of particle size revealed that the diameters of the isolated exosomes varied from 50 nm to 120 nm, and no significant difference in diameter was observed between the exosomes obtained from the healthy individuals and those from patients with lung cancer [Figure 2d]. However, qPCR results showed that the concentration of miR-5703 in serum exosomes from the patients with lung cancer was significantly elevated compared with that in serum exosomes from the healthy individuals (P < 0.0001), [Figure 2e].
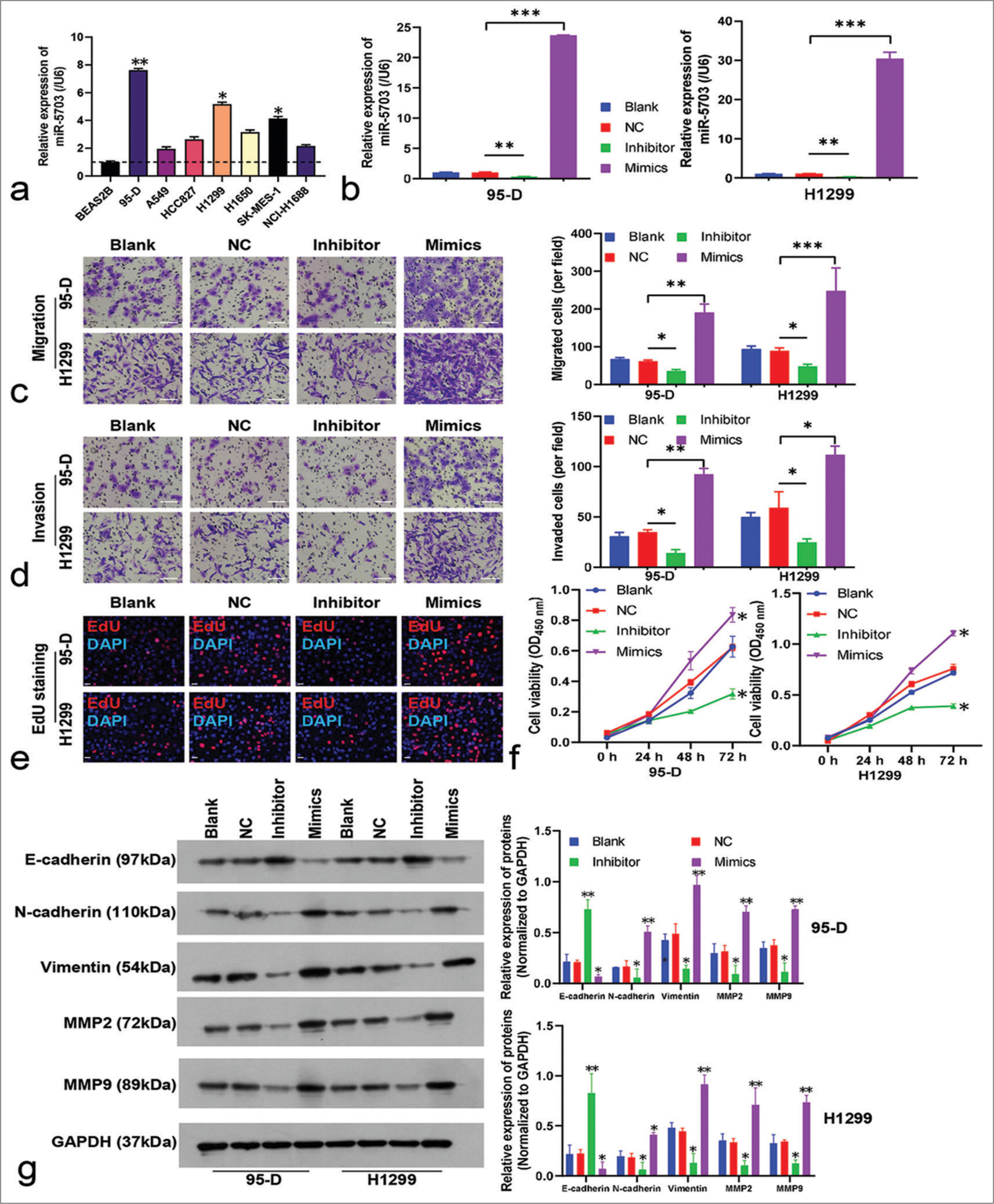
Biological role of miR-5703 in lung cancer cellsTo explore the biological role of miR-5703 in lung cancer, the experiment incorporated various lung cancer cell lines and a non-tumorigenic cell line (BEAS2B). The highest expression levels of miR-5703 were observed in the 95-D and H1299 cell lines [Figure 3a]. Thus, these cell lines were selected for further experiments.
Export to PPT
In the 95-D and H1299 cells, the expression of miR-5703 was manipulated. As shown in Figure 3b, the inhibitors led to a significant decrease in miR-5703 expression (P < 0.01), whereas the mimics caused a dramatic increase in miR- 5703 expression (P < 0.001), [Figure 3b]. Subsequently, the malignant behavior of the cells was assessed. As shown in Figure 3c and d, the inhibitors led to a significant decrease in 95-D and H1299 cell migration and invasion (P < 0.05), whereas the mimics led to an increase in migration and invasion (P < 0.05), [Figure 3c and d]. The results of EdU staining and CCK8 kit on the 95-D and H1299 cells showed that cell proliferation was significantly inhibited and promoted, respectively (P < 0.05), [Figures 3e and f, S1a].
Western blot results showed that the inhibitors enhanced the expression of E-cadherin and suppressed the expression of N-cadherin, vimentin, MMP2, and MMP9. Meanwhile, cells treated with the mimics showed reduced E-cadherin and increased N-cadherin, vimentin, MMP2, and MMP9 expression levels [Figure 3g].
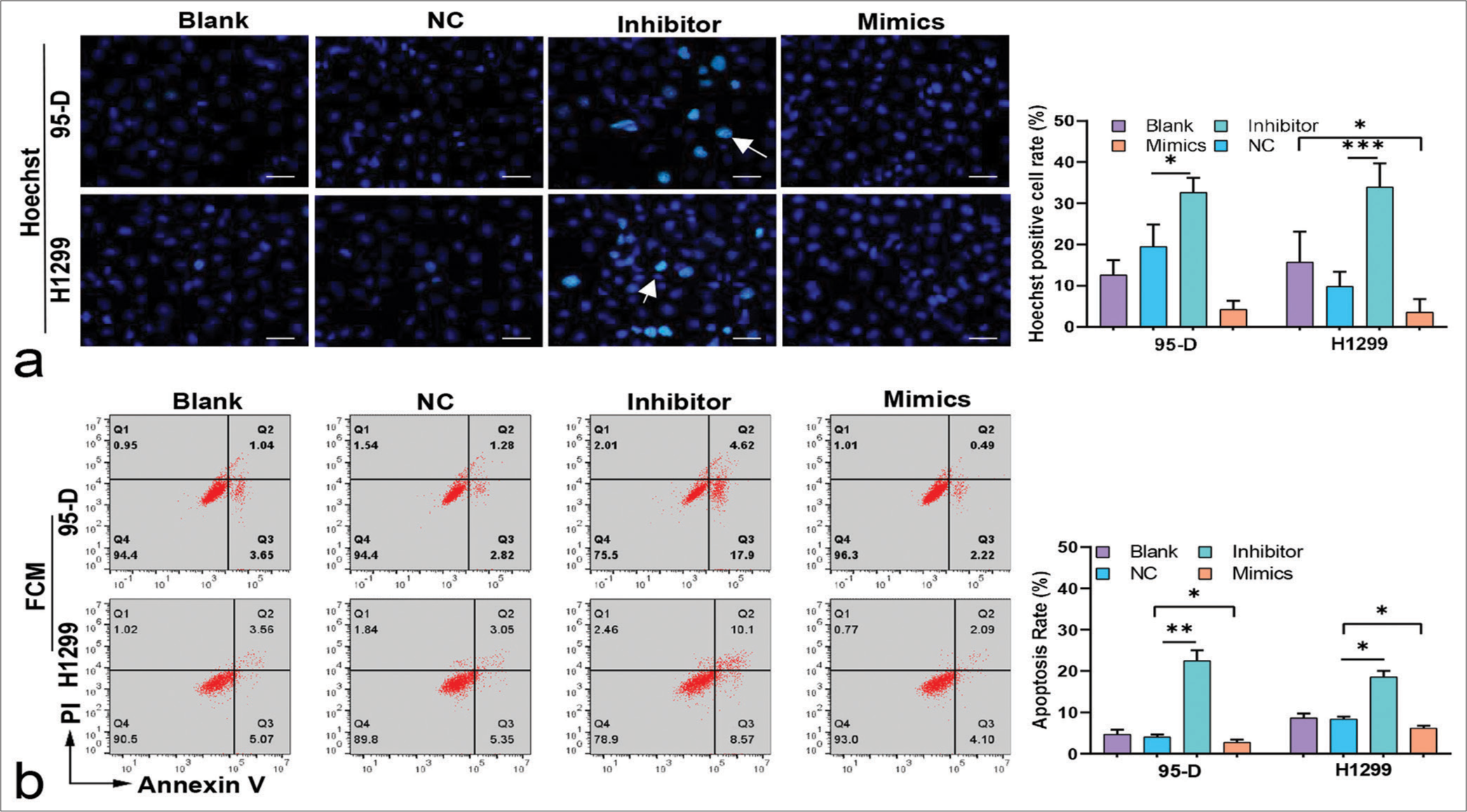
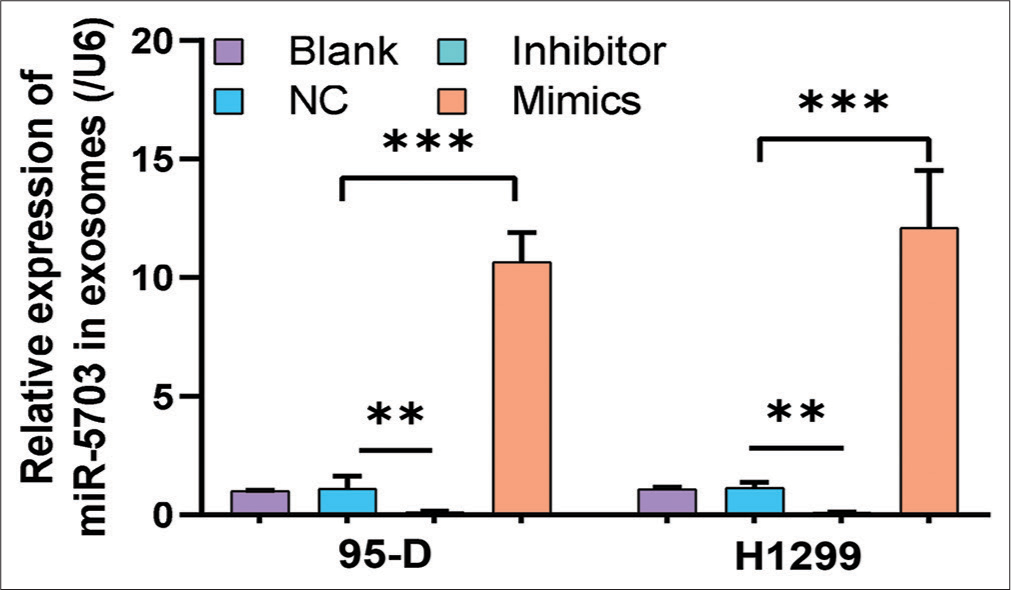
Consistent with the FCM results, Hoechst staining results showed that lung cancer cells treated with the inhibitors had increased number of apoptosis cells (P < 0.05), [Figure 4a and b]. Conversely, after the cells were treated with the mimics, apoptosis rate decreased significantly (P < 0.05), [Figure 4a and b]. These results suggested that miR-5703 promoted the malignant biological behavior of the lung cancer cells. Notably, after treatment with the miR-5703 inhibitors and mimics, the exosomes showed a significant decrease (P < 0.01) or increase (P < 0.001) in miR-5703 abundance, respectively [Figure 5].
Export to PPT
Export to PPT
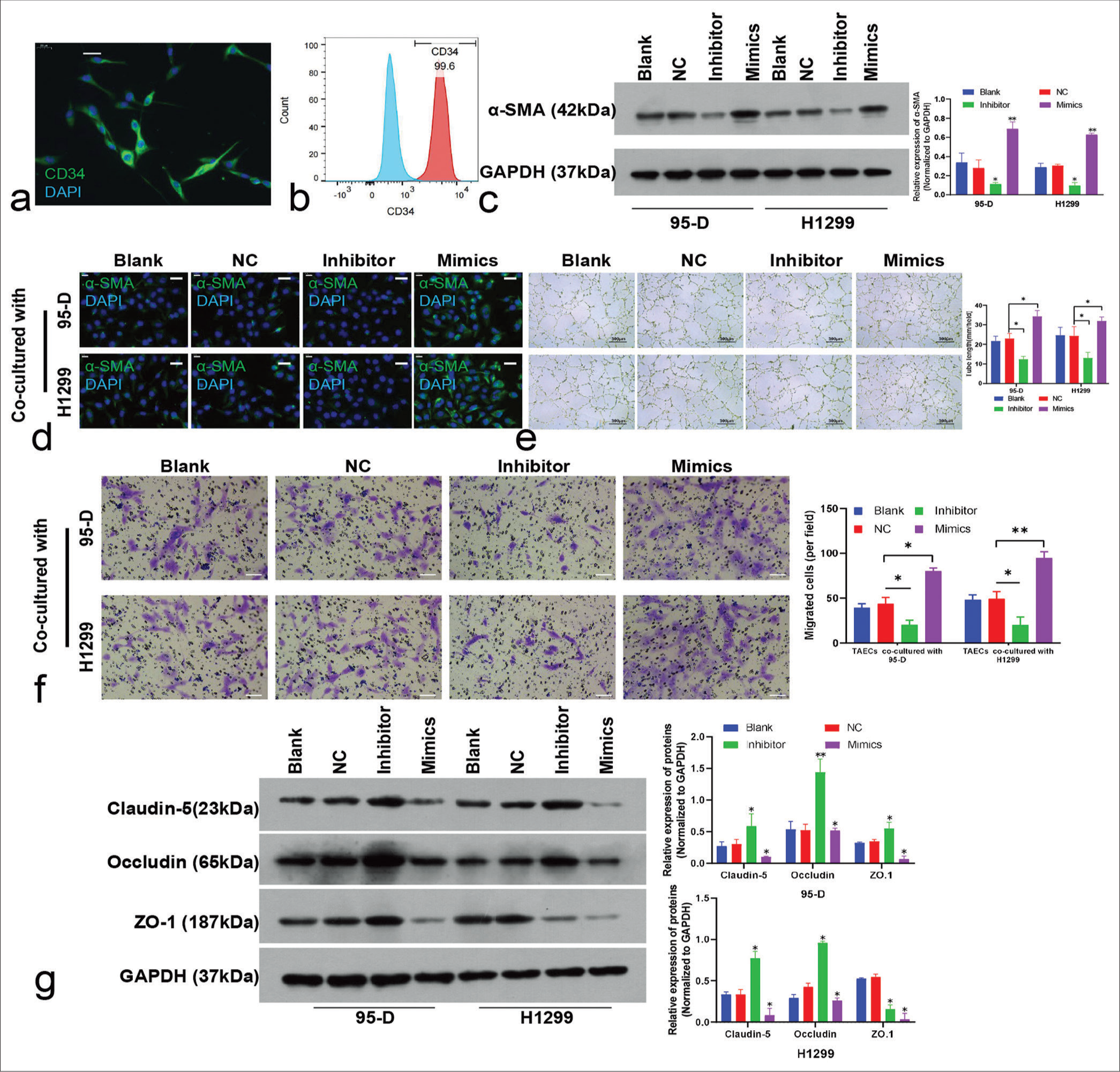
Effect of lung cancer cell–derived exosomes on TAECsTAECs isolated from lung cancer tissues were CD34 positive according to the immunofluorescence [Figure 6a] and FCM results, and the positive ratio exceeded 95% [Figure 6b].
Export to PPT
T
Comments (0)