
Remember me




We obtained samples from PLC patients (n = 22) who underwent surgical resection at Eastern Hepatobiliary Surgery Hospital (EHBH). We selected formalin-fixed paraffin-embedded (FFPE) tissue blocks (n = 24) from 6 patients for proteomics analysis, fresh PLC tissues (n = 25) from 11 patients for ST, and fresh tumor tissues from 5 patients for primary cell isolation (Additional file 1: Table S1). All patients were randomly selected and provided informed consent. We provide the clinical information of these patients, where the presence of cirrhosis and the extent of immune cell infiltration in adjacent liver tissues were determined by two experienced pathologists in a single-blind manner based on HE staining.
Two HCC tissue microarrays (TMAs) were purchased from Shanghai Outdo Biotech Company: Cohort 1 (HLivH180Su17), containing cancerous (n = 92) and adjacent normal (n = 88) tissues, and Cohort 2 (HLivH090Su01), containing leading-edge tissues (n = 84). The clinical and pathological data are summarized in Additional file 1: Table S9.
Mass spectrometry (MS)-based proteomicsMatched tumor and adjacent normal tissues (> 5 mm) were cut into 10 µm thick sections and flattened in preprepared sterilized water. Then, the sections were mounted on Leica PEN Membrane Glass Slides (Cat No. 11505189). The deparaffinization of FFPE tissue sections utilized heptan instead of xylene three times for 15 min, followed by 100% ethanol, 95% ethanol, 70% ethanol, and 50% ethanol (3 min each). After only hematoxylin staining, the FFPE tissue sections were sent for laser-capture microdissection (LCM) by a Leica LMD 7000 (Leica Microsystems, Inc., Bannockburn, IL). All stromal tissues from each tissue section were collected by LCM into a 0.2-ml Eppendorf tube and stored at room temperature for further sample preparation, and the remaining parenchymal tissues were collected into 1.5-ml tubes.
The proteins from FFPE tissue tissues used for MS analysis were extracted by a Qproteome FFPE Tissue Kit (Qiagen) according to the manufacturer’s instructions. Note that since tissue sections had already been deparaffinized in the aforementioned H&E staining step, extraction buffer ExB plus supplemented with β-mercaptoethanol could be directly added to the LCM collection tubes, followed by the steps of extraction and cleanup of proteins from FFPE tissues. Finally, the extracted proteins were subjected to MS analysis after tryptic digestion [39]. Protein expression of the stroma regions and remaining parenchyma regions was analyzed using the data-independent acquisition (DIA) mode of a mass spectrometer (Thermo Fisher). Data-dependent acquisition spectrum libraries were constructed before DIA mode to obtain real spectrum libraries.
Quantitative proteomics analysisThe resulting spectra from each fraction were searched separately against the homo_sapiens_uniprot_2021_3_9.fasta (194,557 sequences) database by the search engine Proteome Discoverer 2.2 (PD 2.2, Thermo). The results of the search and identification by PD2.2 software were imported into Spectronaut (version 14.0, Biognosys) software to generate a library. The eligible peptides and product ions were selected from the spectrum by setting peptide and ion pair selection rules to generate a target list [40]. The DIA data were imported, and ion-pair chromatographic peaks were extracted according to the Target List. The ions were matched, and the peak areas were calculated to qualitatively and quantitatively analyze the peptides. iRT was added to the sample to correct the retention time, and the precursor ion Q value cutoff was set to 0.01. The quantitative values were visualized with Bionic Visualizations Proteomaps (https://proteomaps.net/).
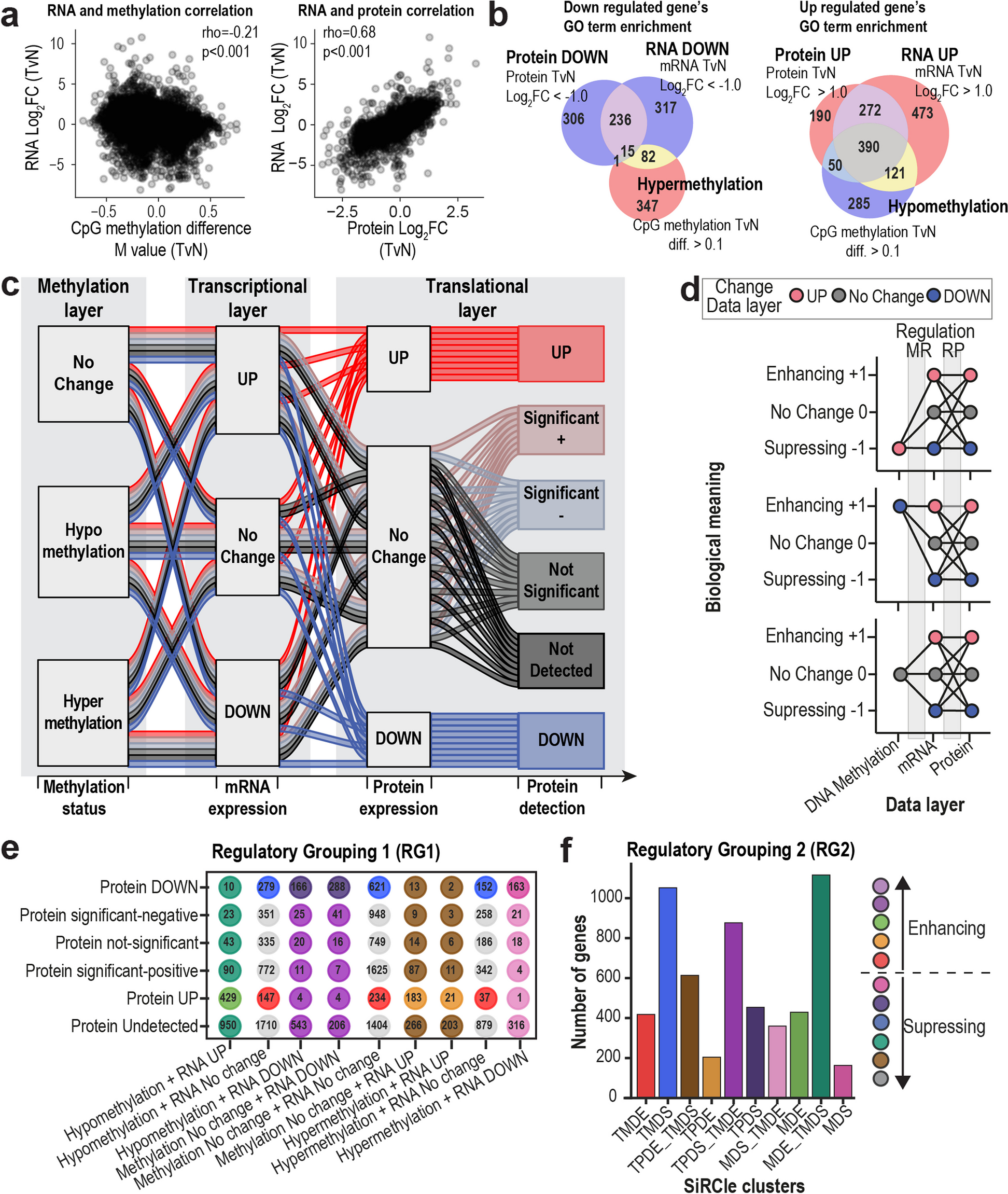
Differentially expressed proteins (DEPs; tumor vs. adjacent nontumor, p < 0.05, |logFC|> 1) underlying enrichment analyses. Gene Ontology (GO) and KEGG pathways with adjusted p < 0.05 (Benjamini‒Hochberg method) were considered significantly enriched.
Spatial transcriptomics experimentsUnder the guidance of pathologists, we defined the interface as the boundary or transitional region between tumor parenchymal cells and adjacent nontumor parenchymal cells (usually 1–2 mm in distance). The definition of interface areas is distinct from that of the capsule, which refers to the fibrous envelope surrounding the hepatocellular carcinoma tumor itself. Each tissue specimen was embedded in optimal cutting temperature compound medium, immediately frozen in an isopentane slurry made with liquid nitrogen, and finally stored at -80 °C until further processing. Each tissue sample was embedded within 30 min for frozen sectioning after surgical removal. The archives of all patients were collected via the EHBH archive system. Informed consent was obtained from the patients, and all procedures were approved by the ethical committee of EHBH.
Spatial transcriptome experiments were performed according to the user guide of the Visium Spatial Gene Expression Reagent Kit (10X Genomics). Cryosections were mounted onto a spatially barcoded array of 10X Genomics Visium with 10-μm thickness. For processing, the tissue was fixed for 30 min with prechilled methanol at − 20 °C, followed by H&E staining. Slides were finally taken on a Leica SCN400 F whole-slide scanner at 40 × resolution. After capturing ideal tissue morphology information and ensuring that RNA was not degraded (RIN ≥ 7), tissue permeabilization and reverse transcription were immediately conducted by a Visium Spatial Tissue Optimization Kit (10X Genomics). Finally, the library was prepared with second-strand synthesis and denaturation and sequenced by NovaSeq 6000 (Illumina). Each of the spots printed onto the array is 55 μm in diameter and 100 μm from center to center, covering an area of 6.5 × 6.5 mm2.
Basis analysis of the spatial transcriptomeData processingThe gene-barcode matrices were analyzed with the Seurat pipeline [41] in R. Spots were filtered for a minimum detected gene count of 200 genes. Then, we used the SCTransform function to perform normalization, log-transformation, centering, and scaling across spots, in which percent.mt and nFeature_Spatial were regressed out in a second nonregularized linear regression. Then, the IntegrateData function was used to integrate the expression data from different sections according to 3000 highly variable genes. The integrated data were used for subsequent clustering.
Differential gene expression and correlation with DEPsDifferential expression analysis was performed using the FindAllMarkers function in Seurat (v.4.0) with the following settings: only.pos = TRUE, assay = "SCT", and slot = "data". Genes filtered for an adjusted p value < 0.05 (Wilcoxon rank sum test, Bonferroni method) were regarded as differentially expressed genes (DEGs) and included in the gene set variation analysis (GSVA).
Next, we compared the correlation between protein and mRNA abundance changes. Spots from seven of these 11 patients with similar clinical features and stromal-rich sections were included. Protein‒mRNA pairs were matched with gene IDs. For these protein‒mRNA pairs, we calculated the Spearman correlation of tumor/non-tumor log2(fold change) for DEPs and DEGs.
Expression heterogeneity between ST spotsIf ρ is the pairwise Pearson’s correlation coefficient between two expression vectors of highly variable genes, the heterogeneity score can be defined as Pearson’s pairwise distance:
We calculated the heterogeneity score between paired spots from the ST data. Then, hierarchical clustering of Pearson’s pairwise distance showed different functional clusters within tumor stroma (T–S) and non-tumor stroma (N‒S). We also calculated the mean Pearson’s pairwise distances for 100 cases sampled in T–S and N‒S. After 500 bootstrap resampling iterations, we fit the distribution and calculated the significance of the difference between the mean pairwise distance in 2 groups (Wilcoxon rank sum test; ref [42].).
Identification of the major cell types for ST spotsTwo methods were used to determine the cell type enriched in each spot: (1) cluster- and marker-based annotation of cell types and (2) estimation of cell purity by deconvoluting cell mixtures from single-cell references by SPOTlight [43].
1)We combined unsupervised clustering and differential expression to compare the top-ranked DEGs with known cell type-specific expression in the literature. In detail, we input the expression profile of the highly variable genes, principal component analysis (PCA) was performed to project the spots into a low-dimensional space, and uniform manifold approximation and projection (UMAP) was performed for visualization. Clustering was performed using the FindClusters function with a resolution of 0.5 to generate 13 clusters, and DEGs were identified in each cluster. Three of these clusters highly expressed hepatocyte-associated genes, such as ALB, SLC10A1, SLC22A1, CYP2A6, and CYP2C8, and were therefore merged and inferred to be hepatocyte cells. Cancer cells are made up of four clusters and highly expressed GPC3, CDC34, and AFP. Cluster 4 highly expressed MS4A1, CD19, CD3E, CD3G, CD8A, CD8B, and CD28, which means that these spots include T cells and B cells. Other cluster markers highly expressed markers specific for cholangiocytes (KRT19, KRT7), fibroblasts (COL1A2, COL3A1, COL1A1, ACTA2), natural killer cells (NKs; NCAM1) and macrophages (CD68, FCGR2A, CD80, CD86, TGFB1, CD163, MRC1). The remaining three clusters had ambiguous cell types, so they were isolated and reclustered, finally obtaining 4 categories assigned to malignant cholangiocytes, T/B cells, and malignant hepatocytes.
2)Single-cell references were downloaded from GSE156625 [44], containing broad cell type annotations from 14 liver cancer patients and 1 healthy donor. To detect marker genes, cells were filtered for a minimum detected gene count of 1500 and were randomly sampled from 100 endothelial cells, fibroblasts, lymphocytes, hepatocytes, and myeloid cells. Then, we used the FindAllMarkers function in Seurat (v.4.0) [41] with the following settings: only.pos = TRUE, min.pct = 0.7 and logfc.threshold = 1. Marker genes and single-cell RDS objects were input into the R package SPOTlight [43] to decompose the expression matrix inferring the percentage of each cell type (cell purity) within a spot. When we focused on the specific cell type, we filtered out those spots that did not have the highest percentage of annotated cell type.
3)Cell type annotation for cohort two: We identified anchors to transfer data from Cohort 1 to Cohort 2 using Seurat’s TransferData function [41]. Each spot obtains a corresponding score for each category, and the category with the highest score is defined as the phenotype of the spot. We examined the cell type annotations in each cohort 2 cluster. We found that the annotation of a small number of cells was inconsistent with that of the majority of cells in the same cluster and corrected their annotation to that of the majority of cells.
Trajectory inference analysisUsing our previously aligned bam files, we first sorted samples by cell barcode. To annotate the spliced and unspliced reads, we used the Velocyto pipeline [45] to generate our loom files. Our steady-state gene-specific velocities were computed and preprocessed following the scVelo python package [46].
Fibroblast subtypes, spatial distribution, and interactionIdentification of fibroblast subtypesWe reclustered fibroblast spots/cells from the ST and scRNA-seq data and then compared their DEGs to match the clusters. For the ST data, 5803 fibroblast-enriched spots were isolated from the broad cell type. Their expression profiles were first combined and the batch effect was corrected using the batch index as a covariate in Seurat [41], then were dimensionally reduced with PCA and clustered at a resolution of 0.5. Similarly, we reclustered 1800 fibroblasts at a resolution of 0.2 from the scRNA-seq data [44] after discarding the cells that expressed fewer than 200 genes or had ambiguous marker genes. We focused on subtypes that were significantly enriched in tumor tissues (Fisher test, p < 0.05).
We mapped clusters in scRNA-seq data to those in ST by scoring single-cell module scores. The list of genes in the module consisted of the top (less than 60) DEGs, and module scores were calculated by three methods (AUCell [47], z score, Garnett [48]). The results of the three methods for assigning single-cell cluster labels to ST clusters were similar, which we showed in Fig. 3D by z scores.
Marker genes and classification of F5-CAFsAfter matching the scRNA-seq and ST data, the common CAF cluster was annotated as F5-CAF because of five marker genes: COL1A2, COL4A1, COL4A2, CTGF, and FSTL1. These five genes were selected from marker genes shared by F5 (from ST data) and TAF4 (from scRNA data). The criteria were q < 0.05 (t test; F5 vs. non-F5 populations), log2(fold change) > 0.5, at least 80% of subtype cells express the gene and other subtypes do not widely express the gene (more than 10% difference).
Based on the expression of the marker genes COL1A2, COL4A1, COL4A2, CTGF, and FSTL1, we employed a random forest model with feature selection to identify the minimal set of genes that could classify F5-CAFs and other fibroblasts. We used this optimized gene set for multiplexed immunofluorescence (mIF) experiments. The ST profile of 80% of the fibroblast-enriched spots was used as the training dataset, and the remaining 20% was used as the test dataset. The classifier was built using a random forest classification method from the randomForest (v.4.7) [49]R package with balanced samples and default parameters. Feature selection was performed by the varImpPlot function to measure variable importance, followed by forward selection to add features. The receiver operation curve (ROC) was plotted by the pROC (v.1.18) [50]R package. Finally, different ROC curves were compared by the roc.test function, and gene combinations with the highest area under the curve (AUC) were selected for F5-CAF classification.
Definition of F5-CAF score and F5-CAF spotWe defined F5-CAF scores as (1) the enrichment score of 5 marker genes of F5-CAFs by ssGSEA [51] and (2) directly summing the normalized expression values of the 5 genes. The results of the two methods are significantly positively correlated. To ensure methodological consistency in module score calculations in subsequent analysis (Fig. 5), we used ssGSEA [51] to calculate the F5-CAF score.
We observed some nonfibroblast-enriched spots with high F5-CAF scores, and there were indeed a few fibroblasts in these spots, as confirmed by morphological features in H&E slides. Although these spots had significantly lower numbers of fibroblasts and thus were not previously defined as fibroblast-enriched spots, they were important in spatial analysis because F5-CAF signatures were also present in these spots. Therefore, when we defined F5-CAF spots, we discretized F5-CAF scores using the median F5-CAF score from F5 spots. This approach preserved the spatial localization of F5-CAFs with as few omissions as possible.
Validation of F5-CAFs by mIF stainingThe TMA was purchased from Outdo Biotech company (Shanghai, China) with clinical information. MIF was performed using OpalTM chemistry (PerkinElmer, Waltham, USA) with five antibodies against EPCAM (Abcam, ab7504), COL4A2 (Abcam, ab125208), CTGF (Abcam, ab5097), FSTL1 (Abcam, ab71548), and COL1A2 (Abcam, ab96723). Briefly, after deparaffinization, The TMA slides were blocked with antibody diluent for 10 min at room temperature after antigen retrieval buffer. The slides were incubated with the primary antibody for 60 min and subsequently incubated with the HRP-conjugated secondary antibody for 10 min after removing the primary antibody and washing in TBST buffer. Thereafter, the slides were incubated with OpalTM working buffer for 10 min at room temperature and then washed in TBST buffer. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain all nuclei.
Panoramic multispectral scanning of slides was performed by the Tissue-FAXS system (TissueFAXS Spectra, TissueGnostics). Then, we imported the data into Strata-Quest analysis software. We used the spectral library for spectral splitting to obtain a single-channel fluorescence signal, The DAPl channel was used to segmentate and identify the effective nucleus. Each of the individually stained tissue spots in the TMA was utilized to establish the spectral library of the fluorophores to eliminate an interference of cross-fluorescence combined with the spectral unmixing algorithm. For organizational area division, a tissue segmentation algorithm from the software was applied to divide each tissue into parenchymal and stromal areas.
Associations between F5-CAFs and prognosisThe association between F5-CAFs and prognosis was analyzed by The Cancer Genome Atlas (TCGA) gene expression and mIF protein expression data. The expression data and clinical data of 371 liver cancer patients in the TCGA datasets were downloaded from cBioPortal. Considering that the gene combination of COL4A2, CTGF, and FSTL1 best discriminated F5-CAFs, we defined risk scores for these three genes:
where Exp is the expression level of each gene, and β is its regression coefficient obtained from the single-variate Cox model. The TCGA-LIHC patients were divided into high-risk and low-risk groups based on the median risk score.
For the mIF data, we counted stromal cells with positive expression of CTGF, COL4A2, and FSTL1 in tumors as F5-CAFs in HCC. Patients were divided into two groups according to the number of F5-CAFs, and the overall and disease-free survival rates in these two groups were compared using Kaplan‒Meier curves and log-rank tests in the R package survminer and survival (https://github.com/kassambara/survminer).
Spatial distribution of F5-CAFsTo examine how the F5-CAF score was influenced by the cancer cells in the tissue samples, we calculated the correlation of the F5-CAF score expression level and its distance to the tumor border.
Masks of the tumor border annotated by pathologists were created in ImageJ software. The mask covered all pixels considered to belong to this specific area. If p is the pixel, all pixels belonging to the mask will form a set Mt. Then, the coordinates of each spot were extracted from the ST data object using Scanpy (v.1.8.2) [52]. The distance from a spot s to the area t is represented by d(s,t), which is defined as the minimal Euclidean distance from the center of spot s to any pixel p from Mt.
$$d\left(s,t\right)=}__}d(s,p)$$
Once distances were determined, for a F5-CAF score of spot s, a tuple (d(s,t), score) was formed. To demonstrate the dependency of the F5-CAF score on the distance to the tumor border, this set of distance-CAF score tuples was then visualized in downstream analysis. To better capture general trends in the data, we used locally weighted scatterplot smoothing (LOWESS) to generate smoothed estimates, which would serve as an approximation of a function f such that score = f(d(s,t)), to be interpreted as if the F5-CAF score value was a function of the distance to the tumor border (statsmodels v.0.31.1 in Python) [53].
Colocalization between F5-CAFs and other cell typesSpatial colocalization analysis was performed in the following two ways:
1)The NeighborRhood R package [54] was used to check whether the coordinates of the spots were significantly enriched in the same area. We input a neighborhood graph containing spot index pairs for adjacent spots and a data frame with spot index and spot phenotype as defined above. Colocalization scores between and within cell phenotypes were calculated for each spot with its neighbors. Then, colocalization scores were compared to a random distribution with shuffling labels using individual one-tailed permutation tests.
2)A new strategy for cell state colocalization:
Cell-state enrichment scores for each spot were calculated by UCell [55], singscore [56], and ssGSEA [51]. Module signatures for 9 cancer cell states and 25 immune cell states were obtained from previous studies [57, 58], representing the typical characteristics of different functional cell subtypes.
First, we compared cell-state scores among three locations: spots near CAFs, spots near fibroblasts, and distant spots (Wilcoxon rank-sum test). The test results were integrated by robust rank aggregation (RRA; https://github.com/chuiqin/irGSEA/), and the significantly different cell states are marked with asterisks in the heatmap.
Second, we defined “Niche intensity” s(i) for each CAF spot i to represent the intensity of a certain state that occurs around the CAF. The niche intensity s(i) for each CAF spot i was defined as the maximum cell-state score of phenotypes of interest (e.g., cancer, immune cells) observed among adjacent spots j ∈ Mi, where |Mi|⩽6.
$$s\left(i\right)=}__}CS(j)$$
The correlation of niche intensity with the F5-CAF score \(cor(s\left(i\right), CS\left(i\right))\) represents the relationship of a specific cell state to the characteristics of F5-CAFs, thereby reflecting the colocalization of F5-CAFs with specific functional cells.
Ligand‒receptor-mediated interactions between F5-CAFs and other cell typesCellphoneDB Statistical Analysis v.2.0 [59] was used to conduct receptor‒ligand analysis between different spot phenotypes as previously described [60]. We input the count matrix from ST data into CellphoneDB and performed the analysis separately for adjacent, interface, and tumor areas. F5-CAF-specific and top significant interactions compared to other fibroblasts were visualized.
Ligands that regulate CSCs were predicted with NicheNet [61]. DEGs of F5-CAFs and their surrounding CSC spots were input to screen the ligands expressed in CAFs and the corresponding receptors expressed in CSCs as potential interactions. CSC spots were selected based on the median of the stemness scores of cancer cell spots around the F5-CAFs. A total of 65 spots with three scoring algorithms exceeding the threshold were identified as CSC spots (1.48% of all cancer cell spots, consistent with the expected proportion of CSCs). Ligand activity ranking was performed according to the previously obtained differentially expressed gene sets. The top 40 ligands were used to obtain downstream activated target genes.
Next, we analyzed the spatial proximity between ligands and receptors [62]. The average of all ligand‒receptor pairs on each slide was first calculated by averaging the ligand and receptor expression between each F5-CAF spot and its six nearest neighbors and then taking the average of the 6 spots. This calculation for each ligand‒receptor pair was then performed on 1000 randomized permutations of spot identities while preserving the total number of spots per replicate to generate a null distribution for each patient. The p value was calculated by the number of randomized permutations that exceeded the true average.
Isolation of F5-CAFs and related in vitro experimentsIsolation and cell culture of CAFsCAFs were isolated from patient tumor tissues, which were stored in 5 mL of MACS Tissue Storage Solution (Miltenyi) and transferred to the laboratory at low temperature. Tumor tissues were washed three times in phosphate buffered-saline (PBS) supplemented with 3% penicillin and streptomycin. Next, the tissues were excised into approximately 1 mm3 pieces and then digested with 0.1% collagenase/dispase (Roche), 0.01% hyaluronidase (Yuanye), and 0.002% deoxyribonuclease (DNase) I (Roche) at 37 °C for 1.5 h until the single cells were approximately 80% confluent. After enzyme digestion, the tissue suspension was filtered through a 70 μm cell screen to remove undigested tissues.
The cell filtrate was collected into a new tube and centrifuged at 1000 rpm for 5 min. The supernatant was discarded, and the cell pellets were resuspended in DMEM/F-12 (Corning) supplemented with 20% fetal bovine serum (FBS) and 1% penicillin and streptomycin, plated in 10 cm culture dishes, and maintained at 37 °C and 5% CO2. When cells were attached for 48 h, they were washed with PBS to remove nonadherent cells, and half of the medium was replaced with fresh DMEM/F-12 supplemented with 10% FBS and 1% penicillin and streptomycin. The process was replaced every 3 days until the cells reached 80 ~ 90% confluence and expanded to passage. The first to fifth passages of CAFs were used in these experiments.
Cell cultureThe human HCC cell lines Huh7 and Hep3B, the human hepatic stellate cell line LX-2, and the human embryonic kidney cell line HEK293T were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin and maintained at 37 °C and 5% CO2.
Immune cell fluorescenceCAFs, LX2, and HEK293T cells were cultured in 96-well cell culture plates until they were attached and fixed with Immunol Staining Fix Solution (Beyotime) for 10 min, washed three times with PBS, permeabilized with Enhanced Immunostaining Permeabilization Solution (Beyotime) for 10 min, and blocked with QuickBlock Blocking Buffer for Immunol Staining (Beyotime) for 10 min. Then, the cells were incubated with primary antibodies (see below) overnight at 4 °C. Subsequently, the cells were incubated with anti-rabbit IgG (H + L), F(ab')2 Fragment (Alexa Fluor 555 Conjugate, CST) or anti-mouse IgG (H + L) Cross-Adsorbed Secondary Antibody (Alexa Fluor 488, Invitrogen). DAPI was used to stain the nuclei. The F5-CAF ratio in CAFs was determined using a Four Color Multiplex Fluorescent Immunostaining Kit following the manufacturer’s instructions (Absin Bioscience). Images were acquired and analyzed with a LionheartTM FX automated Live Cell Imager (BioTek, USA). The primary antibodies used were alpha-smooth muscle actin (α-SMA) (mouse monoclonal, Boster, BM0002, 1:50), fibronectin (FN) (CST, 26,836, 1:200), COL1A2 (Abcam, ab96723, 1:500), COL4A2 (Abcam, ab125208, 1:1200), CTGF (Abcam, ab5097, 1:700), and FSTL1 (Abcam, ab71548, 1:700).
Supernatant collection of CAFsCAFs and LX2 cells were seeded in 10 cm dishes in 10 ml of DMEM supplemented with 10% FBS and 1% penicillin and streptomycin until 80 ~ 90% confluence. Thereafter, fresh medium was added, and the conditioned supernatant was collected 48 h later and centrifuged at 2000 rpm for 5 min to remove nonadherent cells and cell debris. All supernatants were frozen at − 80 °C until further use.
Cell proliferation assayHuh7 and Hep3B cells were seeded at a density of 200 cells/well in 96-well plates. The experimental groups and control group cells were cultured in 200 μL of the aforementioned supernatants and DMEM supplemented with 10% FBS. Then, cell viability was measured by a Cell Counting Kit-8 (CCK-8) (NCM) at 6, 24, 72, and 120 h, and the absorbance of the cells at 450 nm wavelength was detected using the Agilent Synergy H1 Microplate Reader (BioTek, USA).
Coculture experiments and interference assayDirect and indirect coculture methods were used for the coculture experiments in this study. In brief, CAFs and LX2 cells were mitotically inactivated in DMEM supplemented with 8 μg/mL mitomycin C (Rhawn) at 37 °C with 5% CO2. The cells were incubated for 2 h and washed three times with PBS. Then, they were used as feeder layers for coculture. Subsequently, direct coculture was performed by seeding feeder layer cells (700/well) first and then Huh7 (500/well) or Hep3B (50/well) cells in the upper chamber of 24-well plates. Indirect coculture was performed by seeding Huh7 (500/well) and Hep3B (50/well) cells in the lower chamber and feeder layer cells (700/well) in the upper chamber in a 24-well Transwell apparatus with a 0.4 µm pore size (Corning). Huh7 and Hep3B cells were cultured in a medium supplemented with 10% FBS and 5% calf serum, respectively, for further analysis.
The feeder layers of CAF5 and CAF9 cells were used for the interference assay. Based on the aforementioned coculture experiments, two neutralizing antibodies against discoidin domain receptor 1 (DDR1) (CST, 5583) and COL4A2 (Abcam, ab125208) were used. The antibodies were used at a dilution of 1:2000.
Colony formation assayCocultured cells were grown for approximately 10 days, and colonies were fixed with 4% paraformaldehyde and stained with crystal violet for assessment. Colonies containing > 50 cells were counted under a microscope.
Quantitative real-time polymerase chain reaction (RT‒qPCR)Total RNA was extracted from cultured Huh7 or Hep3B cells using RNAiso Plus (Takara), and the RNA concentration (ng/mL) and purity were measured using a Nanodrop 2000 spectrophotometer (Thermo Scientific, USA). RNAs with OD260/OD280 ratios ranging from 1.8 to 2.1 were used in the following experiments. The FastKing RT Kit (with gDNase) (Tiangen) was used to reverse transcribe the total RNA. RT‒qPCR was performed using FastFire qPCR PreMix (SYBR Green) (Tiangen) according to the manufacturer’s instructions. Glyceraldehyde-3 phosphate dehydrogenase (GAPDH) was used as the reference gene to normalize the mRNA levels. Data were collected with a LightCycler 480 instrument (Roche), and the mRNA levels were analyzed as log2 of the fold difference. The primer sequences are listed in Additional file 1: Table S12.
Statistical analyses were performed using GraphPad Prism 8 software. One-way analysis of variance (ANOVA) and two-way ANOVA were used for multiple comparisons. The error bars in the experiments indicate the standard error of the mean or standard deviation for a minimum of each experiment in triplicate. P < 0.05 was considered statistically significant.
Comments (0)