Pathogenic variants in the OTOF gene, encoding the membrane-associated protein otoferlin, are the most common cause of auditory neuropathy spectrum disorders (ANSD), accounting for 1–8% of recessive, non-syndromic sensorineural hearing loss (SNHL), DFNB9 [1, 2]. Otoferlin is a key calcium ion sensor protein involved in inner hair cell (IHC) signaling to auditory nerve fibers, transmitting acoustic signals with high temporal accuracy needed for auditory perception, including sound localization and speech comprehension [3]. OTOF pathogenic variants lead to presynaptic ANSD reflected in normal otoacoustic emissions (OAE) and/or normal cochlear microphonics (CM), indicating proper outer hair cell function, while the auditory brainstem response (ABR), is absent or abnormal, indicating a characteristic phenotype of hearing loss with severe impaired speech discrimination. The phenotype is typically characteristic by severe-to-profound SNHL with congenital or prelingual onset or by atypical mild-to-moderate SNHL that might be temperature-sensitive or progressive [2].
The Deafness Variation Database (https://deafnessvariationdatabase.org/gene/OTOF) includes 197 pathogenic and 26 likely pathogenic variants, including at least 35 splice variants, known to cause autosomal recessive OTOF-related ANSD with varying prevalence. Many of these are founder alleles among different ethnic groups [4]. Significant phenotypic differences, including type of audiogram, severity of hearing loss and onset, were observed, with most biallelic loss of function (LoF) variants resulting in more severe SNHL compared to other variant combinations [5].
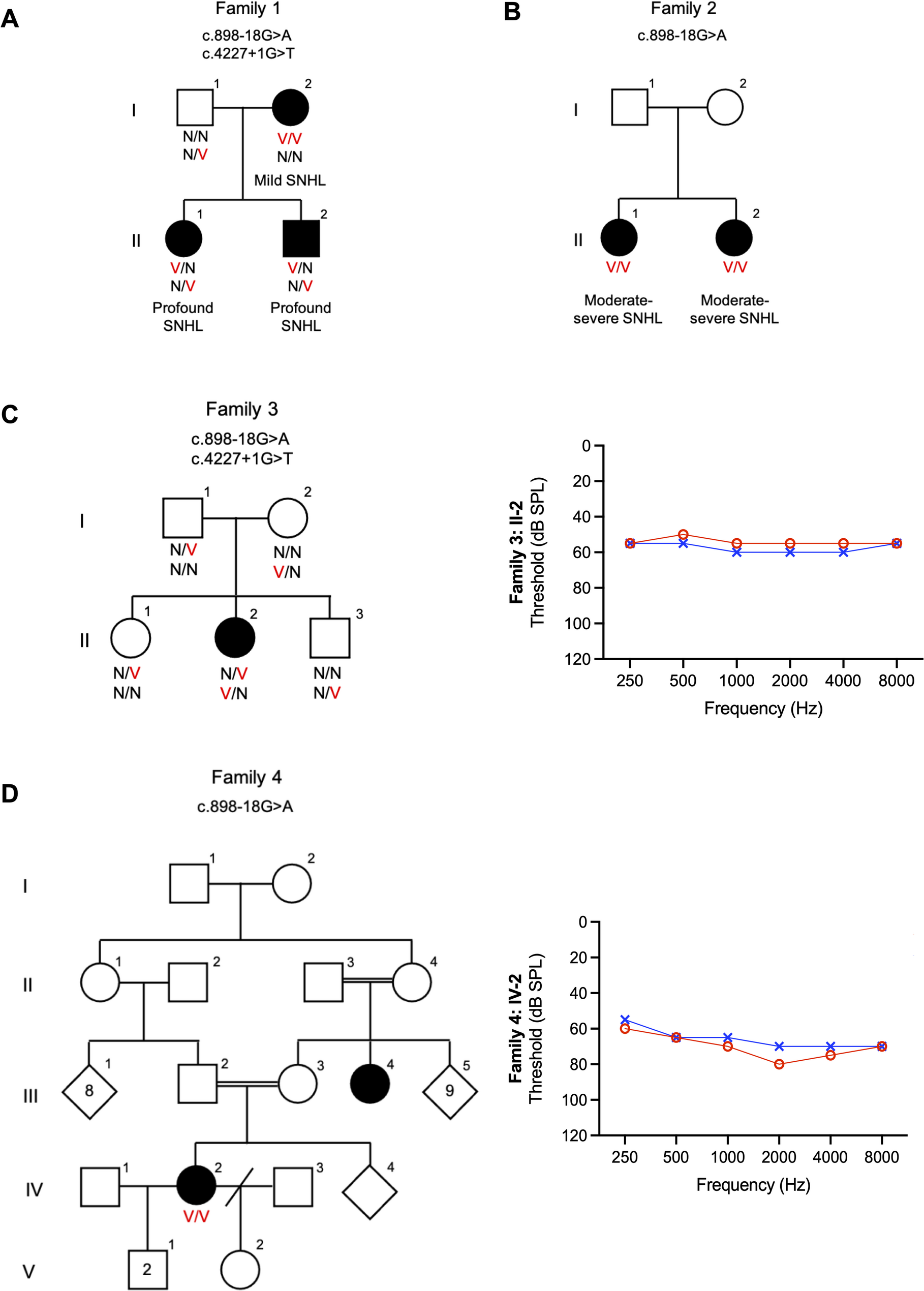
Founder pathogenic variants are known to increase the prevalence of diseases in closed ethnic groups, including hearing loss and ancestry-specific pathogenic variants in different Jewish communities [6]. In data collected from 24 populations across the world, 56 deafness-causing founder mutations were reported in 14 known deafness genes, including GJB2, GJB6, GSDME, TMC1, TMIE, TMPRSS3, KCNQ4, PJVK, OTOF, EYA4, MYO15A, PDZD7, CLDN14, and CDH23 [7]. In Israel, deafness-related founder mutations were detected in 18 genes: CDH23, CEACAM16, COL4A3, COL4A5, GJB2, GJB6, LOXHD1, MYO3A, MYO7A, MYO15A, OTOF, PCDH15, SLC26A4, SYNE4, TMC1, USH1C, USH2A and USH3A [6, 8,9,10,11,12].
According to the Israeli Medical Genetic Database (https://medicine.ekmd.huji.ac.il/en/imgd/Pages/Search_by_ethnic_group.aspx), there are 323 disease founder variants in the Jewish population, 35 of which are founder variants in the Moroccan Jewish population, including the most common deafness variant in this population, TMC1 c.1939T > C:p.(Ser647Pro), with a carrier rate of 5.7%, and accounting for 34% of hearing loss in this community, turning the TMC1 gene into the second most prevalent deafness gene in the Jewish population, after GJB2 [10]. In this study, we identified a novel splice variant, c.898-18G > A, in the OTOF gene [NM_001287489.2; Chr2:26712626 C > T (GRCh37); Chr2: 26489758 (GRCh38)], segregating with ANSD in seven Jewish Moroccan families from five medical centers in Israel, with a carrier rate of 1.5% and no carriers detected in other Jewish ethnicities. This is the second founder variant in the Moroccan Jewish population associated with hearing loss.




Comments (0)