
Remember me


Nanotube Scaffolds (NTS) were synthesized by electrochemical anodization at room temperature. For this purpose, titanium foil (Advent Research Materials Ltd., 0.1 mm thickness, 99.6+ % purity) was cut into small pieces of 20 mm x 30 mm, cleaned with distilled water and isopropyl alcohol for 10 min each in an ultrasonic bath, and then dried in a nitrogen stream. The cleaned titanium foil was used as the anode and a platinum mesh as the cathode. Both electrodes were clamped in a holder with a fixed distance of 45 mm. The setup was immersed in an electrolyte (98% ethylene glycol, 2% distilled water) containing 0.3% or 0.6% ammonium fluoride for single or double anodization, respectively. For single anodized NTS, 0.3%-NH4F-electrolyte was used, and a voltage of 50 V was applied for 60 min, resulting in free-standing nanotubes with a tube diameter of 72 ± 3 nm. 0.6%-NH4F-electrolyte was used to synthesize double anodized NTS. The first anodization step takes 30 min at a voltage of 50–60 V [20]. At this production point, we produced single anodized nanotubes with a diameter of about 70–100 nm, respectively. The TiO2 nanotubes were then completely removed from the Ti-sheet by ultrasonication in distilled water followed by drying in a nitrogen stream. The pretreated Ti-sheet was subjected to a second anodization step for 30 min at a voltage of 50–60 V, resulting in a thin interconnected nanoporous surface layer a few nanometers thick, with underlying nanotubes. The tube diameter of the nanoporous layer is 72 ± 2 nm and 102 ± 5 nm, respectively. NTS were allowed to rest overnight in ethylene glycol. Finally, to remove any residual electrolyte, the scaffolds were cleaned with distilled water by ultrasonication for 10 min. Note: We tested additional tube diameters, single and double anodization, but for the sake of simplicity we have limited the description to the tubes shown in Fig. 1.
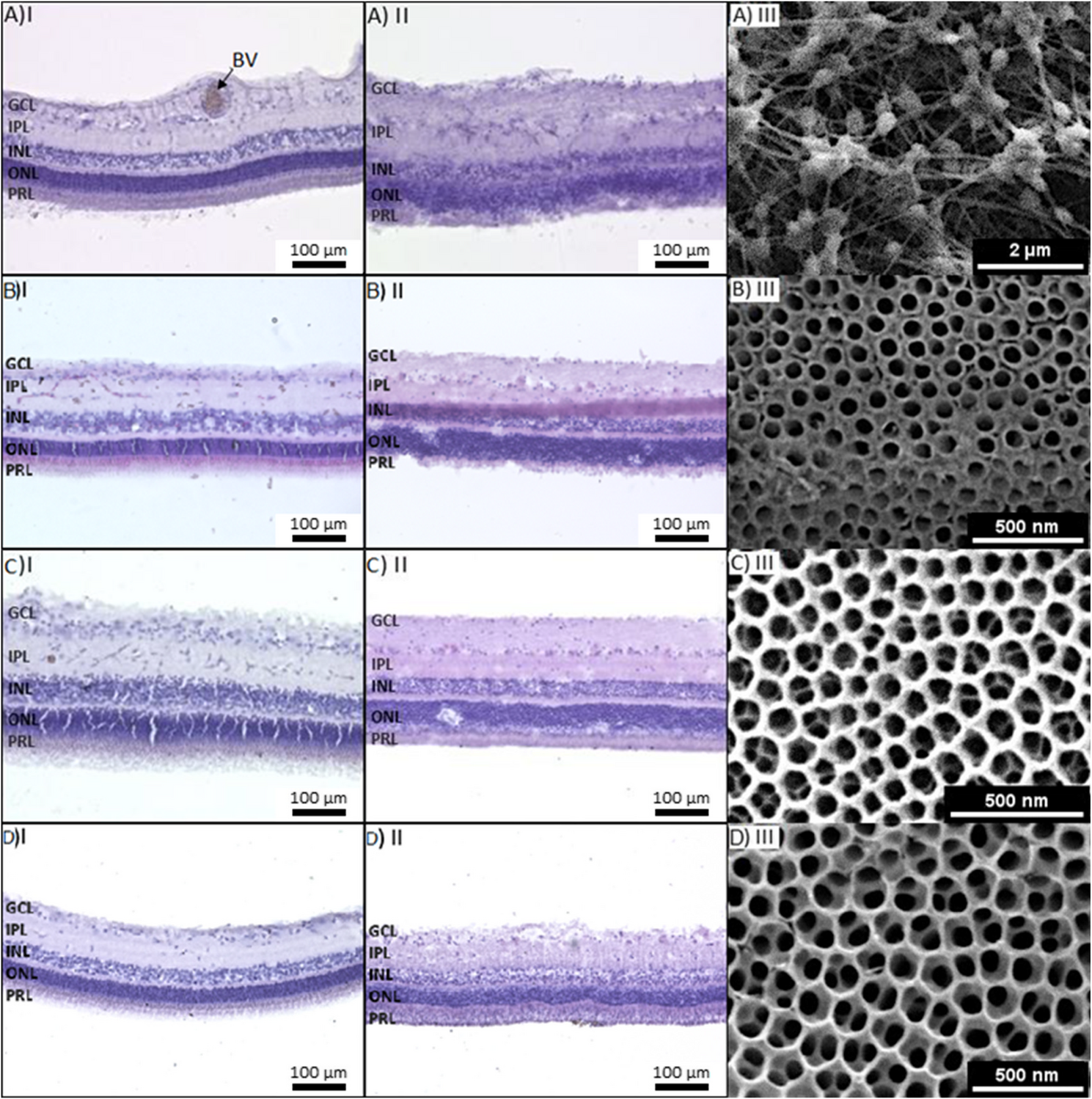
Fig. 1
Organotypic culture of adult vascularized porcine retina explants on different scaffolds. Hematoxylin and eosin staining of a porcine retina before (I) and after (II) culture on different substrates for 5 days. Column (III) shows the corresponding cultivation substrate: A porcine retina cultured on Millicell® filter, B single anodized nanotube scaffolds with tube diameters of 72 nm, C double anodized nanotube scaffolds with tube diameters of 72 nm and D double anodized nanotube scaffolds with tube diameters of 102 nm. After culturing, structure and tissue integrity are clearly visible for the double anodized nanotube substrates, but not for the Millicell® filters or single anodized nanotube scaffolds. The layers of the retina are not maintained, indicating a loss of tissue integrity and properties of different layers. However, very good structural preservation (especially of the photoreceptor segments) was found on both double anodized nanotube scaffolds. BV: blood vessel, GCL: ganglion cell layer, INL: inner nuclear layer, IPL: inner plexiform layer, ONL: outer nuclear layer, PRL: photoreceptor segment layer
Retina Preparation and Organotypic Culture of Porcine RetinaAdult retina from porcine eyes was kindly provided by the slaughterhouse Emil Färber GmbH Großschlächterei & Co. KG, Belgern Schildau, Germany. Pigs were slaughtered at the age of 6 to 7 months and weighted approximately 100 to 150 kg. After individual electrocution and subsequent exsanguination, pig eyes were enucleated by a qualified butcher, leaving 3–5 mm of the optic nerve intact. Eyes were transferred to ice-cold PBS (4–8 °C, pH 7.4) and transported to the laboratory. Transportation took about 2 h. The eyeball was cleaned of surrounding tissue such as muscle, fatty tissue and connective tissue, transferred to a tube filled with 70% ethanol for some seconds to sterilize the ocular surface, washed twice in sterile PBS. The eye was opened along the ora serrata under sterile conditions in warmed culture medium. After disconnecting the vitreous body, the retina was always punched square (13 mm x 13 mm) centrally below the optic nerve. The quadratic punched samples were always taken from the same origin or position of the eye, separated from their surroundings, detached from the retinal pigment epithelium and carefully placed with the photoreceptor side down on the NTS and on the Millicell® cell culture inserts (hydrophilic PTFE-membrane, pore size 0.4 μm, PICM0RG50, Merck Millipore) for comparison purposes. Porcine retina was cultured using the air-liquid interface method as follows.
Therefore, NTS was placed on a sterile, stainless-steel grid (30 mm x 30 mm x 5 mm) inside a culture dish (Ø = 5 cm). The culture medium (Advanced DMEM/F-12, 0.1% gentamycin, 10% horse serum, 2% Glutamax) was added, so that the medium was in contact with the bottom of the scaffold but did not overflow it. This ensures a free gas exchange.
For cultures using the Millicell® inserts, the retina was placed on an insert with the photoreceptor side down and inserted in a culture dish (Ø = 5 cm). Culture medium was added until the filter was wet, but not submerged. The cultures were incubated at 37 °C and 5% CO2 for 5 days.
Afterwards, the retina was fixed on top of the NTS or the filter with fixation solution (4% PFA in PBS, pH 7.4) for at least 4 h or overnight. Due to the strong adhesion of the tissue to the NTS, the tissue was carefully removed from the NTS surface with a razor blade. On the Millicell® inserts the adhesion was weak, so they could be removed easily by a brush. The fixed tissues were then processed for histological staining (HE and immunohistochemical staining).
Histological Processing (hematoxylin/eosin Staining, HE) and Brightfield Microscopy30 μm slices of porcine retina of gradual post mortem times were stained with hematoxylin/eosin (HE) method to visualize tissue morphology from time point of enucleation at the slaughterhouse (post mortem hour 0) until post mortem hour 6. This experimental setting was chosen to simulate different transportation duration from slaughterhouse to bench, which we will call “post mortem time” in the following. During this time, the eyes were kept in ice-cool PBS solution. These time ranges were chosen to determine potential effects of post mortem ages on retinal tissue degradation and consequently on cultivation success any potential limit of post mortem preparation times. Therefore, the pig eyes were kept in ice-cool PBS from the time of enucleation until appropriate post mortem time and were then prepared as described above for NTS culture, but once the retina was isolated, it was not cultivated on NTS, but fixed in 4% PFA (in PBS, pH 7.4).
Fixed retinal samples were washed in PBS (pH 7.4) and embedded in 3% agarose. Samples were cut into 30 μm thin tissue slices using a vibratome and stored in PBS-NaN3 at 4 °C until further processing for staining. Slices for post mortem analysis were stained with hematoxylin and eosin (HE). Therefore, slices were briefly rinsed in PBS, transferred to distilled water and mounted on a microscope slide. The slides were then placed on a heating plate (35 °C) until evenly dried and fixed. After desalting the slides in a cuvette filled with distilled water, hematoxylin was applied for 4 min. Slides were then washed three times with water within 30 min, before eosin was applied for 4 min. The slides were then immersed in distilled water, then in 80% ethanol and finally in distilled water again until the agarose was clear. Excess water was removed, the slides dried on a heating plate and immediately covered with mounting medium on a coverslip. Slides were stored upright, at room temperature and protected from light for 24–48 h before microscopic analysis using a Keyence fluorescence microscope (BZ-9000, Keyence, Neu-Isenburg, Germany).
Vital Dye Staining of Living Retinal Tissue Ex Vivo and Fluorescence MicroscopyAll steps were performed at room temperature. Living (non-PFA-fixed) retinal wholemounts were explanted and mechanically fixed on Whatman Filters with their vitread surface up, and placed into extracellular solution (136 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 MgCl2, 10 mM HEPES, 11 mM glucose), adjusted to pH 7.4. The wholemounts were loaded with vital dyes resolved in extracellular solution: 1 µM MitoTracker Orange (chloromethyltetramethylrosamine, M-7510, Invitrogen), CellTracker Green (5-chloromethylfluorescein diacetate; C-2925, Invitrogen) and FM™ 1–43 (Invitrogen). After appropriate incubation time, the wholemounts were transferred to a dye-free, extracellular solution filled chamber and examined using a confocal laser scanning microscope LSM 510 (Zeiss, Oberkochen, Germany) by viewing from the vitread side. The following cell membrane-permeant thiol-reactive vital dyes were tested: See Fig. 2, A-E; II and III.
Fig. 2
Morphology and viability of porcine retina at 0–6 h post mortem, symbolizing the effect of long transportation times from slaughter house to bench. Column I: hematoxylin/eosin staining of porcine retinas PFA-fixed at 0, 1, 3, 4 and 6 h post mortem. Column II: isolated living retinas incubated with in vivo fluorescent dye CellTracker Green (CTG), Müller glia cells were exclusively CTG positive and appear in green. Nerve fibers and ganglion cells somata (AII) appear black displaying no fluorescent staining. In E II a branched blood vessel is recognizable. Column III: retinas incubated with in vivo fluorescent dye MitoTracker Orange (MTO) and FM1-43 (green). MTO staines exclusively Müller cells and intact Müller cells/Müller cell end feet appear in orange. FM1-43 staines nerve fibers/ganglion cell axons and the cell membrane of ganglion cell somata. The distribution pattern of these in vivo fluorescent dyes are very specific and display the status of oxidative stress in the isolated retinal tissue. In E III (arrows) some ganglion cell somata were filled with FM1-43 showing the first damage of the integrity of the cell membrane due to oxidative stress. GC: ganglion cells, MC: Müller glial cells, NF: nerve fibers, BV: blood vessel
Immunohistological Staining and Fluorescence MicroscopyPorcine retinae was cultivated on two types of NT scaffolds (tube diameter 72 nm ± 3 nm and column 102 nm ± 5 nm) at 37 °C for 5 days. Retinae were fixed with 4%-PFA over night and carefully removed from NT scaffold. For immunohistological processing the tissue was embedded in 3% agarose (in PBS, pH 7.4). Samples were cut into 30 μm thin tissue slices using a vibratome and transferred into well-plates for staining process. The following steps were performed by using PBS (pH 7.4) + 1% DMSO + 3% triton-X-100, for incubation and washing. The slices were incubated in 5% donkey normal serum (Jackson Immunoresearch) for 1 h at room temperature and then incubated with primary antibody overnight at 4 °C (1:500 anti-GFAP by Dako; 1:100 anti-GαT1 by Santa Cruz; 1:100 anti-GαT2 by Santa Cruz, 1:500 anti-Glutmanine synthetase (GS) by Merck Millipore; 1:500 anti-IBA by Wako; 1:100 anti-PNA by Sigma; 1:500 anti-S100β by Sigma, 1:100 anti-Vimentin/V9 by Dako). After washing the slices 3 times for 1 h each, the fluorescent secondary antibodies (1:200, Jackson Immunoresearch) and Hoechst33358 (H3570, 1:1000, Life Technologies) were added and incubated for 2 h at room temperature and protected from light. Slices were washed, fixed and mounted on a microscope slide and stored upright until microscopic analysis using a confocal laser scanning microscope LSM 510 (Zeiss, Germany).
Comments (0)