Bioinformatics Analysis
RNAseq data for pan-cancer and corresponding normal tissues from TCGA and GTEx were downloaded from UCSC XENA website (https://xenabrowser.net/datapages/). The GSE10810 and GSE42568 datasets were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The 3D structure diagram of USP53 was generated online using SWISS-MODEL (https://swissmodel.expasy.org/). The GEPIA2 website (http://gepia2.cancer-pku.cn/) contains RNAseq data for 9736 tumors and 8587 normal tissues as well as provides the corresponding python package for visual analysis. The Kaplan-Meier Plotter (http://kmplot.com/) online tool can assess the expression of all genes and their correlation with survival in more than 30,000 samples from 21 cancers.
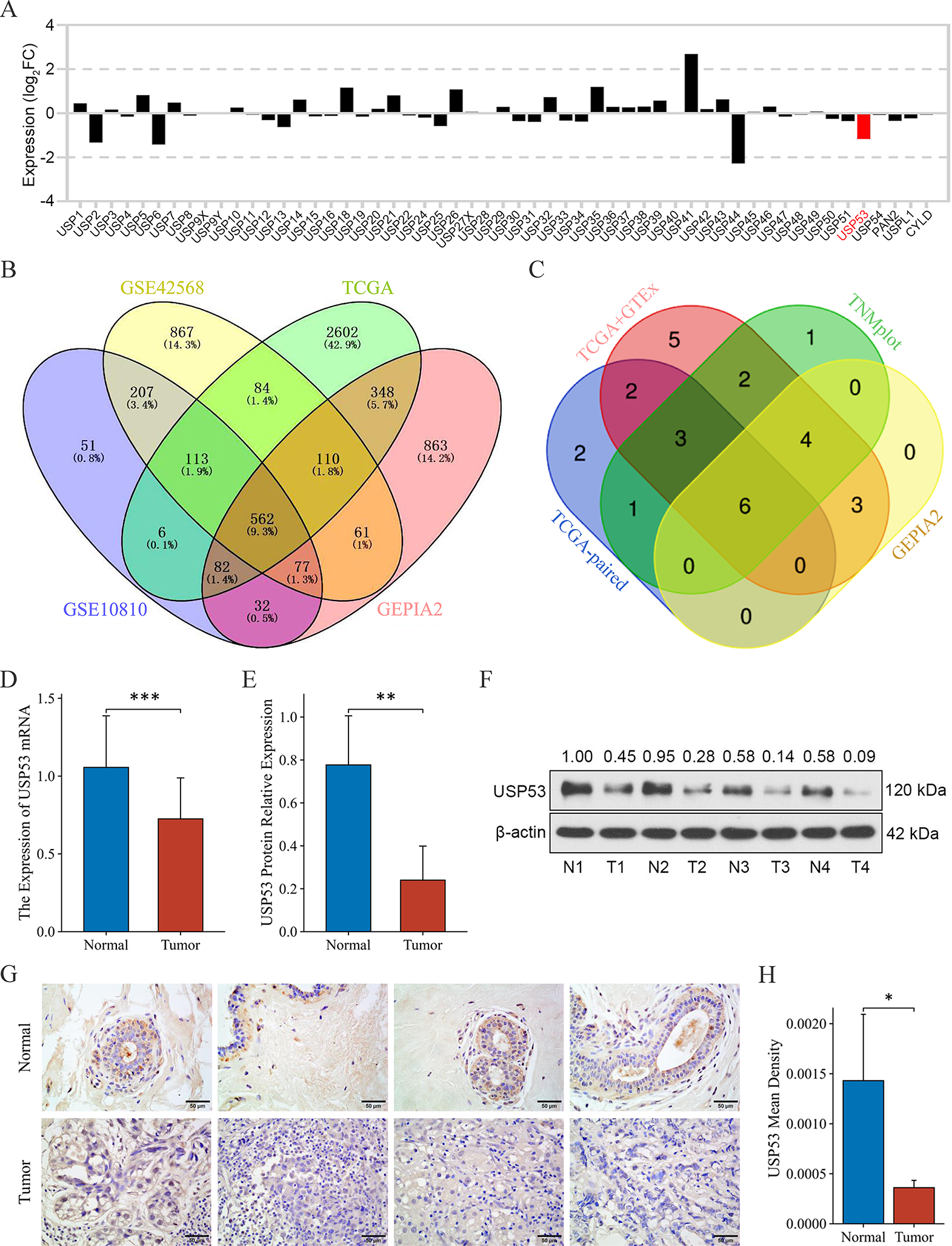
The Limma package in R software was used to analyze the differential expression of USP family members in breast cancer and normal breast tissue from the TCGA database. Differentially expressed genes in breast cancer from GSE10810, GSE42568, TCGA and GEPIA2 were screened respectively, with the condition as log2FC < -1 and padj < 0.05, then take the intersection of the four to obtain the down-regulated gene set.
GEPIA2 and Kaplan-Meier Plotter were used to analyze the differences of USP53 expression in the corresponding pan-cancer database. Stats and Car package was used to analyze USP53 expression differences and paired expression differences between 33 cancers and normal tissues from TCGA and GTEx databases. The results of the above four analyses were summarized to obtain the intersection cancer species.
The correlations between USP53 and all genes in breast cancer was analyzed by Pearson method, and then the conditions of |Cor| > 0.5 and padj < 0.05 were set to screen for USP53 correlated gene sets. The GO&KEGG enrichment analysis of the selected gene sets was performed using the ClusterProfiler package. The pairwise similarity of the enriched items was calculated by Jaccard similarity index, and then cluster analysis was performed by Hclust function.
The diagnostic value of USP53 in breast cancer was analyzed using the pROC package. The Kaplan-Meier Plotter online tool was used to analyze the survival differences betwween patients with high and low expression of USP53/ZMYND11.
The Ggplot2 and VennDiagram packages were used to visualize the data analysis results above.
Collection of Clinical Tissue Samples
Thirty pairs of fresh breast cancer and para-carcinoma tissues, and seventy-three paraffin breast cancer specimens, from November 2022 to October 2023, were collected from the Breast Disease Diagnosis and Treatment Center of First Hospital of Qinhuangdao. The breast cancer was diagnosed with pathological examination, and the tissues were removed with surgery. The paired breast cancer and para-carcinoma tissues were used for PCR and western blot detections, and the paraffin specimens were applied for immunohistochemistry staining. The informed consent was obtained from every patient. Experimental procedures were conducted according to the Declaration of Helsinki, which was approved by the Ethics Committee of Qinhuangdao First Hospital (approval number: 2022k009).
Construction of Plasmid
To investigate the effect of USP53, its coding sequence was cloned into pcDNA3.1 vector, and the silencing fragment targeting USP53 was inserted into pRNA-H1.1/Neo vector. To explore the function details of USP53, its mutant sequence with 33–50 amino acid residues deficiency was synthesized and inserted into pcDNA3.1 plasmid. To verify the interaction between USP53 and ZMYND11, the USP53 coding sequence was inserted into pcDNA3.1-flag vector, and ZMYND11 coding sequence was inserted into pcDNA3.1-HA vector. The knockdown fragment targeting ZMYND11 was also inserted into pRNA-H1.1/Neo vector to suppress its expression. The USP53 or ZMYND11 overexpression plasmids were purchased from YouBio (Changsha, Hunan, China), and the knockdown sequences were synthesized by General Biol (Chuzhou, Anhui, China). The silencing sequences targeting USP53 or ZYMND11 were shown below:
shNC: GTTCTCCGAACGTGTCACGTTCAAGAGAACGTGACACGTTCGGAGAATTTTT
shUSP53-1: GGGATATCAGTGGTGTTAAATTCAAGAGATTTAACACCACTGATATCCTTTTT
shUSP53-2: GGGAAAGATGTTGTCTCCAATTCAAGAGATTGGAGACAACATCTTTCCTTTTT
shZMYND11: GGGCTATAGATCTTAATAAATTCAAGAGATTTATTAAGATCTATAGCCTTTTT
Cell Culture and Treatment
Human immortalized mammary epithelial cell lines (MCF-12 A) and breast cancer cell lines (MCF-7, T47D, BT474, SKBR3, MDA-MB-453, MDA-MB-231) were purchased from iCell (Shanghai, China). MCF-12 A was cultured with the special medium provided by the manufacturer. MCF-7, T47D, BT474 and SKBR3 were cultured with MEM (SolarBio, Beijing, China). MDA-MB-453 and MDA-MB-231 were cultured with L-15 medium (Servicebio, Wuhan, Hubei, China). 10% FBS and 1% penicillin + streptomycin were added to MEM and L-15. The cells were grown at 37 C in a humidified atmosphere of 5% CO2.
Transfection was performed using LipofectamineTM 3000 reagent (Invitrogen, Carlsbad, CA, USA) in serum-free medium. The cells with transfection of USP53-overexpressed or -silenced vector were treated with G418 (500 µg/ml for MCF-7 cells and 600 µg/ml for MDA-MB-231 cells; Biosharp Life Science, Hefei, Anhui, China) at 24 h post transfection for 1–2 weeks, and the single cells were selected for continuous culture with G418 for 2 weeks. The surviving cells were considered as USP53-stably-overexpressed or silenced cells.
To intercept the protein synthesis, a translational inhibitor cycloheximide (CHX) (20 µg/ml; Aladdin, Shanghai, China) was applied, and a proteasome inhibitor MG132 (10 µM; Macklin Inc, Shanghai, China) was used to block protein degradation.
Real-Time PCR
The concentration of total RNA was measured with the NANO 2000 ultraviolet spectrophotometer (Thermo Scientific, Wilmington, DE, USA) after being extracted from tissues or cells using the TRIpure Total RNA Extracting Kit (BioTeke, Beijing, China). BeyoRT-II M-MLV reverse transcriptase (Beyotime Institute of Biotechnology, Shanghai, China), with random primer as RT primer, was used to reverse transcribe RNA into cDNA (1 µg RNA from cells or 5 µg RNA from clinical specimens for one repeat). The reagent and instruments used in RT were RNase-free. Subsequently, the real-time quantificational PCR was performed to measure the expression of USP53, in presence of 2×Taq PCR Master Mix and SYBR Green (SolarBio), with 1 µl cDNA as the template. PCR procedure was set as follows: 95 ℃ for 5 min 10 s, 60 ℃ for 10 s, 72 ℃ for 15 s, followed with 40 cycles of 72 ℃ for 1 min 30 s, 40 ℃ for 1 min, melting 60–94 ℃, every 1 ℃ for 1 s, and finally incubation at 25 ℃ for several min. For detection of one marker, three technical repeats were set for one experiment, and three individual experiments were performed. The data was calculated with 2−ΔCt or 2−ΔΔCt method. The primers were synthesized by General Biol, and the sequences were shown in the following, USP53 forward: 5’-TTATCAGCCTGGAAGTAT-3’; USP53 reverse: 5’-GCATCTCCCTGACAAAC-3’; β-actin forward: 5’- GGCACCCAGCACAATGAA − 3’; β-actin reverse: 5’- TAGAAGCATTTGCGGTGG − 3’.
Western Blot
Western and IP lysis buffers (Beyotime Institute of Biotechnology, Shanghai, China) supplemented with phenylmethanesulfonyl fluoride (1 mM) were used for protein extraction. After concentration determination, the protein was separated with SDS-PAGE (20 µl protein for one lane), and transferred onto polyvinylidene fluoride membrane (Abcam, Cambridge, UK), which was then blocked for 60 min, and incubated with the following primary antibodies at 4 ℃ overnight in the dark: rabbit anti-USP53 (1:500; cat. No. A14353, Abclonal, Shanghai, China), rabbit anti-ZMYND11 (1:1000; cat. No. GTX103403, GeneTex, Inc., Irvine, CA, USA), and mouse anti-β-actin (1:1000; cat. No. sc-47,778, Santa Cruz Biotechnology Inc. USA). As soon as the membrane was rinsed, goat anti-rabbit or anti-mouse secondary antibodies were incubated at 37 °C for 45 min with HRP (1:5000; Beyotime Institute of Biotechnology), followed by ECL reagent interaction and signal exploration. Gel-Pro-Analyzer software was used to analyze the optical density of the bands. β-actin served as the internal control.
Immunohistochemistry Staining
The tissues were made into routine paraffin sections, which underwent deparaffination with xylene and ethanol, and antigen retrieval at boiling. Afterwards, 3% H2O2 was used to eliminate the peroxidase, and 1% BSA was used to block the non-specific antigen. Incubation with primary antibody against Ki-67 (1:50; cat. No. AF0198, Affinity, Cincinnati, OH, USA) was performed at 4 ℃ overnight, and that of secondary antibody labeled with HRP was executed at room temperature for 60 min. Subsequently, the sections interacted with DAB reagent, stained with hematoxylin, soaked with 1% hydrochloric acid/ethanol, dehydrated with ethanol and xylene, finally mounted with gum.
The staining results were scored according to a previous report [24]: percentage of immunoreactive cells: 0 (0–5%), 1 (6–25%), 2 (26–50%), 3 (51–75%) and 4 (76–100%); and staining intensity: 1 (negative), 1 (weak), 2 (moderate) and 3 (intense). USP53 expression was scored by multiplying intensity and percentage. Statistically, a staining score below 6 represents low expression, and a score above 7 represents high expression.
CCK-8 Assay
In 96-well plates, cells were treated with CCK-8 reagent (10 µl per well) for two hours. After that, the optical density of the supernatant was determined using a microplate reader at 450 nanometers.
Colony Formation Assay
The MCF-7 and MDA-MB-231 cells with stable expression of USP53 were used for colony formation assay. The cells were seeded in culture dishes with 300 cells in per dish, and culture for about two weeks. The medium was refreshed every three days. After culture for two weeks, the cell clones in dished were fixed and stained with Giemsa stain (Jiancheng Bioengineering Institute, Nanjing, China) for 5 min, and the clone numbers were counted.
Flow Cytometry
The flow cytometry was performed to detect the cell cycle and apoptosis. The cells were collected and fixed with 70% ethanol at 4 ℃ overnight. The cells were washed with PBS, incubated with RNase A at 37 °C for 30 min, and stained with propidium iodide (PI) in the dark for 30 min. Then the cell cycle phase was determined by flow cytometer (Agilent, Santa Clara, CA, USA). The proliferative index was calculated based on the cell percentage in each phase: (G2 + S)/G1.
For apoptosis detection, the cells were treated with Annexin V-FITC at room temperature for 10 min in the dark, and then stained with PI for 5 min. Immediately, the determination was performed with flow cytometer.
Measurement of Activity of Caspase-3/9
The activity of caspase-3 and caspase-9 was assessed with Caspase 3 Activity Assay Kit or Caspase 9 Activity Assay Kit (Beyotime Institute of Biotechnology) according to the manufacturer’s protocols. Briefly, the cells were collected and lysed on the ice, and the protein concentration was determined with Bradford Protein Assay Kit (Beyotime Institute of Biotechnology). Afterwards, the sample was incubated with acetyl-Asp-Clu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) substrate at 37 ℃ for 1 h to produce the yellow pNA, and the absorbance at 405 nm was measured with a microplate reader. The standard curve was drawn using standard pNA with grading concentrations, and activity of caspase-3 or caspase 9 was calculated based on the pNA concentration. One unit was the amount of enzyme that will cleave 1.0 nmol of the colorimetric substrate Ac-DEVD-pNA per hour at 37 ℃ under saturated substrate concentrations.
Reactive Oxygen Species (ROS) Determination
The ROS content in cells were determined with a ROS Assay Kit (Biosharp Life Science, Hefei, Anhui, China) according to the manufacturer’s instruction. The collected cells were firstly incubated with dichlorodihydrofluorescein-diacetate (H2DCFDA) reagent (10 µM) at 37 ℃ for 30 min in the dark. Then the cells were washed twice, and the fluorescence intensity in cells was detected using flow cytometer.
Mitochondrial Membrane Potential (MMP) Evaluation
The MMP of cells was evaluated using a Mitochondrial Membrane Potential Detection Kit (JC-1) (Biosharp Life Science). The cells cultured on the glass sides were incubated with JC-1 reagent at 37 ℃ for 20 min, and washed by buffer twice. The images were acquired with a fluorescence microscope at 200× magnification. The MMP ratio was calculated by JC-1 aggregates (red)/JC-1 monomers (green).
Immunofluorescence Staining
Immunofluorescence staining was applied for detection of USP53 and ZMYND11 location in cells. The cells were pre-seeded on glass sides, and fixed with 4% paraformaldehyde (Sinopharm Chemical Reagent Beijing Co., Ltd, Beijing, China) for 15 min, permeated with 0.1% triton-100 (Beyotime Institute of Biotechnology) for 30 min, and blocked with 1% bovine serum albumn (BSA) (Sangon, Shanghai, China) for 15 min. Subsequently, the cells were incubated with antibody against with USP53 (1:100; cat. No. H00054532-B01P) or ZMYND11 (1:100; cat. No. H00054532-B01P, Abnova, Taipei City, Taiwan) at 4 ℃ overnight, and incubated with secondary antibody conjugated with Alexa Fluor® 488 or 555 fluorescent dye (1:200; CST, Boston, MA, USA) for 60 min. Finally, the nuclei were stained with DAPI (Aladdin, Shanghai, China), and sides were mounted with anti-fading reagent (SolarBio). The images were acquired at 400× magnification.
Co-Immunoprecipitation (Co-IP)
Co-IP was applied with a Co-IP Assay Kit (Pierce, Rockford, IL, USA) to verify the interaction between USP53 and ZMYND11, or ZMYND11 and ubiquitin. The antibody was pre-conjugated onto AminoLink resin, and incubated with cells lysate at room temperature for 2 h. After washing, the antigen-antibody complex was eluted and collected for SDS-PAGE according to the previous description. The antibody against HA (cat. No. AE008, Abclonal) or myc tag (cat. No. AE070, Abclonal) was used at 1:500 dilution.
Xenograft Model
Healthy female BALB/C nude mice with six-week-old was kept in a controlled environment (12 h light/12 h dark cycles, 21–23 ℃, humidity of 45–55%) with free access to food and water. The MCF-7 cells with USP53-stable-expression or -knockdown were subcutaneously inoculated in mice with 107 cells per mouse (n = 6 in each group). The tumor size was measured every three days using a vernier caliper. At 21 days post inoculation, the mice suffered euthanasia of 60% CO2 inhalation, and the tumors were isolated for subsequent detections.
The feeding and experiments of animals were carried out according to Guide for the Caree and Use of Laboratory Animals (8th, NIH), and approved by the Ethics Committee of Qinhuangdao First Hospital (approval number: 2022k009).
TUNEL
The tumor tissue was made into conventional paraffin sections. After deparaffination with xylene and ethanol, the sections were permeated with 0.1% triton X-100, and incubated with TUNEL reagent (Roche, Nutley, NJ, USA) at 37 ℃ for 60 min in a humid and dark box. Then the sections were stained with DAPI, and mounted with anti-fading reagent. The images were acquired at 400× magnification.
Statistical Analysis
The data in this study were shown as mean ± standard deviation (SD), and analyzed utilizing R or GraphPad Prism software. The data from two independent groups were compared with unpaired t test or Wilcoxon rank sum test (non-normal or heterogeneity of variance). The data from two paired groups were compared with paired t test or Wilcoxon sign rank test (non-normal). The data in multiple groups were analyzed with ANOVA or Kruskal-Wallis (non-normal or heterogeneity of variance), followed with Bonferroni’s multiple comparisons test. The data from immunohistochemistry staining of breast cancer specimens were analyzed with Chi-square or Fisher’s exact tests. A p value less than 0.05 was considered as statistically significant.



Comments (0)