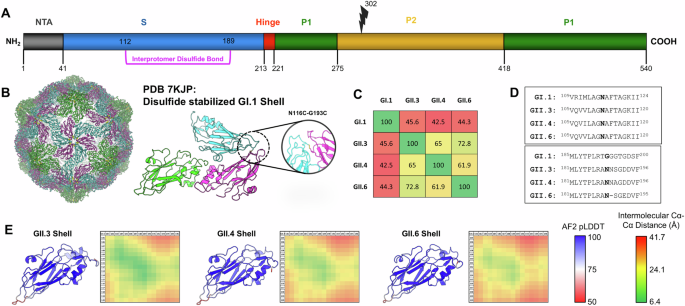
Structural modeling of T = 3 GII.3, GII.4, and GII.6 VP1 shell domains
Monomeric models of the shell domains from VP1 proteins (residues 1–220) were generated with AlphaFold2 using sequences for GII.3 (Genbank Accession MT409884), GII.4 (Genbank Accession MK754446), and GII.6 (Genbank Accession MH260487)34. High-confidence predictions (pLDDT >85) were generated for each shell sequence and three copies were subsequently aligned to the asymmetric unit of the GI.1-DS1 T = 3 shell structure (PDB 7kjp). These T = 3 models were subjected to energy minimization and FastRelax protocols in PyRosetta to remove any clashes that may have been generated during the structure alignment35. Intermolecular distances were measured between all Cα atoms from 2 loops containing residues 105–120 and 181–196 in the modeled T = 3 arrangements. 2D matrices corresponding to these intermolecular distances were plotted as a heatmap for visualization.
Plasmid construction and VLP expression
Protein sequences of norovirus VP1 from genotypes GII.3 (Genbank Accession MT409884), GII.6 (Genbank Accession MH260487) and GII.4 (Genbank Accession MK754446) were mammalian codon-optimized by Genewiz (South Plainfield, NJ) and subcloned into a eukaryotic-expression vector under the control of the CMV promoter. DS1 variants were generated by introducing cysteine substitutions at amino acid positions N112 and N189 within the VP1 protein of each genotype. Caspase cleavage mutants (GII.3-D302A and GII.3-D302S) were generated by introducing alanine or serine substitutions at amino acid position D302 within VP1 of GII.3. Plasmids were transiently transfected into Expi 293 F cells (ThermoFisher, Waltham, MA) using Expifectamine (ThermoFisher, Waltham, MA) following the manufacturer’s recommended protocol. At 72 h post-transfection, a 1 mL sample of each culture was harvested into a microcentrifuge tube and centrifuged at 10,000 × g. Clarified supernatant was decanted into a fresh tube. The cell pellet was solubilized in 0.5 mL lysis buffer, centrifuged at 10,000 × g, and the clarified cell lysate was decanted into a fresh tube. Cell lysis buffer contained 0.5% Triton X-100, 40 mM Tris-HCl, pH 8.0, 120 mM NaCl, and 1X EDTA-free protease inhibitor (Roche Diagnostics, Indianapolis, IN). Clarified supernatant and cell lysate were used for SDS-PAGE and western blot analysis. Remaining bulk cell cultures were harvested 72 h post-transfection and frozen at −70 °C. To generate clarified bulk supernatant, the bulk harvests were thawed at room temperature (RT), mixed thoroughly by inversion, and centrifuged at 3500 × g for 10 min at RT. The resulting clarified bulk supernatant was transferred to a clean, sterile tube and stored at −70 °C.
VLP purification
The frozen bulk cell culture was thawed and clarified through a Sartopure GF+ (Sartorius, Bohemia, NY) depth filter. The clarified VLPs were precipitated using 1.5 M ammonium sulfate (AS) and pelleted by centrifugation at 5000 × g for 10 min. The pellet was resuspended in an anion exchange chromatography (AEX) loading buffer and filtered through a 0.2 µm PVDF membrane (MilliporeSigma, Burlington, MA). The filtered AS precipitation product was purified by AEX using a Sartobind Q membrane (Sartorius, Bohemia, NY) and the VLPs were eluted in an elevated ionic strength buffer. The AEX eluates were further purified by size exclusion chromatography (SEC) using a Sepharose 6 FF (Cytiva, Marlborough, MA) column. Selected SEC fractions were pooled, concentrated, and exchanged into a formulation buffer using a 300 kDa UF/DF membrane (Repligen, Waltham, MA). The UF/DF product was spiked with a cryoprotectant and filtered through a 0.2 µm PVDF membrane (MilliporeSigma, Burlington, MA). All purified VLPs were frozen and stored at −70 °C. The final concentration of the VLPs was 1 mg/mL as measured by the Bradford Assay (ThermoFisher, Waltham, MA) using the manufacturer’s recommended protocol.
SDS-PAGE and western blot
Clarified cell lysate and clarified supernatant were resolved on a 4–12% gradient, 1.5 mm, 15-well Bis-Tris NuPAGE gel in 1X MES buffer (ThermoFisher, Waltham, MA). For SDS-PAGE, where equal protein was loaded, the protein concentration was determined by Bradford assay. SeeBlue Plus2 pre-stained protein standard (ThermoFisher, Waltham, MA) was included on the SDS-PAGE for molecular weight reference. For Coomassie staining, SDS-PAGE gels were rinsed with water, stained with SimplyBlue Safestain (ThermoFisher, Waltham, MA) for 1 h at RT and destained with water at RT overnight. For Western blot analysis, proteins were transferred onto nitrocellulose membranes using the iBlot western blotting system (ThermoFisher, Waltham, MA). Membranes were transferred to the Bandmate Automated Western Blot Processor (ThermoFisher, Waltham, MA) for blocking and probing. Membranes were blocked for 1 h at RT with 30 mL of 5% non-fat dry milk (Blotting-grade blocker, Bio-Rad, Hercules, CA) in 1X tris-buffered saline containing 0.1% Tween-20 (1X TBST). Membranes were probed with a primary antibody cocktail of 2 mouse norovirus VP1 monoclonal antibodies at a dilution of 1:1000 in 5% non-fat dry milk in 1X TBST for 1 h at RT. The primary antibody cocktail consisted of a 1:1 mixture of two mouse norovirus VP1 monoclonal antibodies: My BioSource.com clone # M120539 (MyBioSource.com, San Diego, CA, cat # MBS832466) and Abcam clone # NVGC-01 (Abcam, Waltham, MA, cat # ab272687). Membranes were probed with an alkaline-phosphatase-conjugated affinipure goat anti-mouse IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA, cat # 1115-055-146) diluted 1:1000 in 5% non-fat dry milk in 1X TBST for 1 h at RT. Membranes were washed 3 times for 5 min each at RT with 1X TBST. Membranes were developed using 1-step NBT/BCIP (Pierce, ThermoFisher, Waltham, MA) according to the manufacturer’s recommendations. Coomassie-stained SDS-PAGE gels and western blots were imaged using an Epson Perfection V370 Photo Scanner and Epson Scan Software version 3.9.2.5US.
Sucrose gradient purification
30 mL frozen bulk lysates were thawed in a water bath, clarified by centrifugation, and concentrated to 1 mL using an Amicon 100 kDa MWCO centrifugal filter unit (MilliporeSigma, Burlington, MA). 5 μL of input material was saved for SDS-PAGE analysis. 16 mL, 15–36% continuous sucrose gradients in phosphate-buffered saline (PBS) were made on the day of purification using a Hoefer SG15 gradient maker (Hoefer, Bridgewater, MA) connected to a Cole Palmer Master Flex L/S Digital peristaltic pump flowed at 3.6 mL/min using size 16 tubing capped with a blunt 4-inch 14-gauge needle placed at the bottom of a 17 mL ultracentrifuge tube. 1 mL of concentrated bulk lysate was gently layered on top, and gradients were placed in SW28.1 buckets and loaded into a SW28Ti swinging bucket rotor (Beckman Coulter, Indianapolis, IN). Samples were centrifuged for 3 h at 150,000 × g in a Beckman Optima CL-100K Ultracentrifuge (Beckman Coulter, Indianapolis, IN) operated at 4 °C under vacuum. Gradients were manually fractionated from the top down using a pipette into 12 × 1.34 mL fractions. 15 μL of each fraction was analyzed on a 4–12% SDS-PAGE gel, and norovirus VLPs consistently eluted at the center of the gradient. Fractions 5–7 were pooled and dialyzed overnight using a 25 kDa MWCO dialysis cassette against norovirus VLP storage buffer containing: 25 mM Tris pH 7.5, 150 mM NaCl, 0.02% PS-80, and 5% sucrose. Protein concentration was estimated by Bradford assay using a BSA standard curve, and concentrations were adjusted to approximately 0.5 mg/mL for use in subsequent assays. VLPs were aliquoted, flash frozen in liquid nitrogen and stored at –80 °C.
Dynamic light scattering (DLS)
The hydrodynamic diameter and polydispersity index (PdI) of purified VLPs were measured using a Malvern Zetasizer Nano ZS instrument (Malvern Panalytical, Westborough, MA). Measurements were recorded using 0.1 mL of purified VLPs at 0.5 mg/mL in norovirus VLP storage buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 0.02% PS-80, and 5% (w/v) sucrose) in a ZEN040 cuvette (Malvern Panalytical, Westborough, MA) using the following instrument parameters:
Material = Protein (RI = 1.450, Absorption = 0.001)
Dispersant = 25 mM Tris, 150 mM NaCl, 5% (w/v) sucrose (Viscosity = 1.0454 cP, RI = 1.339)
Temperature = 25 °C
Method = Mark-Houwink
Cell type = ZEN0040
Equilibration time = 60 s
Back scatter = 173⁰
3 measurements each (automatic duration)
General-purpose data analysis was performed in Zetasizer software. Intensity distribution values of the 3 replicates were plotted in GraphPad Prism software for visualization.
VLP treatment
100 mM stocks of diamide (Sigma) and beta-mercaptoethanol (Acros Organics) were freshly prepared in PBS buffer. 2.5 µg of VLPs from each genotype were treated with either diamide or BME at a final concentration of 20 mM at RT in PBS buffer for 1 h. Samples were mixed with 1X LDS buffer (ThermoFisher, Waltham, MA) and boiled for 10 min prior to analysis via SDS-PAGE as described above.
Nano differential scanning fluorimetry (nanoDSF)
Thermal stability assays were performed using a NanoTemper Prometheus NT.48 nanoDSF instrument (Nanotemper, Watertown, MA). Purified VLPs at a concentration of 0.5 mg/mL in norovirus VLP storage buffer were loaded into 3 NanoTemper high-sensitivity capillaries (3 technical replicates each). An initial discovery scan was used to optimize excitation power at 280 nm. Temperature was ramped from 20 °C to 95 °C at a rate of 1 °C per minute, and fluorescence was recorded at 330 nm and 350 nm. The 330/350 fluorescence ratio and its derivative were used to fit the inflection point (Tm). The average 330/350 fluorescence signal of the 3 replicates was plotted in GraphPad Prism software for visualization.
Electron microscopy
Purified VLP samples, along with norovirus VLP storage buffer, were shipped to Nano Imaging Services (NIS, San Diego, CA) for negative-stain electron microscopy analysis. The samples were diluted to a final concentration of 0.012 mg/mL with buffer. Samples were deposited on a layer of continuous carbon supported by nitrocellulose on a 400-mesh copper grid. The grids were prepared by applying 3 μL of sample suspension to a cleaned grid, blotting away with filter paper, and immediately staining with uranyl formate. Samples were imaged on an FEI Tecnai T12 electron microscope (serial number D1100), operating at 120 keV and equipped with an FEI Eagle 4k x 4k CCD camera. Negative-stain grids were transferred into the electron microscope using an RT stage. Data collection is carried out using Leginon software, where high magnification images are acquired by selecting targets at a lower magnification54,55. Images of each grid were acquired at multiple scales to assess the overall distribution of the specimen. After identifying potentially suitable target areas for imaging at lower magnifications, high magnification images were acquired at nominal magnifications of 110,000x (0.099 nm/pixel), 52,000x (0.212 nm/pixel) and 21,000x (0.513 nm/pixel). The images were acquired at a nominal underfocus of −5.0 μm to −1.2 μm and electron doses of ~10–25 e-/Å2. Representative micrographs for each sample collected at 52,000x were selected for qualitative comparisons. All micrographs from the 52,000x magnification were imported into RELION (v4.0.1)56 and CTF corrected using CTFFIND (v4.1.14)57 VLPs were manually picked from micrographs and were extracted in a 300 ×300 pixel (636 × 636 Å) box and subjected to 2D classification using 4 classes and a particle diameter of 500 Å.
Saliva binding affinity assay
Maxisorp 384-well assay plates (Thermo Fisher, Waltham, MA) were coated with 25 μL of human saliva (Precision for Medicine, Frederick, MD) diluted 1:500 in Dulbecco’s phosphate-buffered saline (DPBS). Plates were sealed, centrifuged at 1500 × g for 1 min and incubated overnight at 4 °C in a humidity chamber. A norovirus VLP masterblock was generated by diluting VLPs in 5% non-fat dry milk powder (NFDMP) in phosphate-buffered saline containing 0.1% tween-20 (PBS-T) to a starting concentration of 20 μg/mL, followed by a two-fold, ten-point serial dilution. The masterblock was sealed, centrifuged at 1500 × g for 1 min and stored overnight at 4 °C in a humidity chamber. The assay plates were washed 6x with 1X PBS-T (100 µL/well) for 1 min each and blocked with 5% NFDMP in PBS-T (80 μL) for 30 min in a humidified incubator (21 °C, 80% RH). VLPs were added to 384-well assay plates and incubated for 1 h in a humidified incubator to allow for VLP binding of human saliva (21 °C, 80% RH). Following incubation, the assay plates were washed 6x with 1X PBS-T (100 µL/well) for 1 min each. Saliva-bound VLP was detected using GII.3-specific polyclonal rabbit serum in 5% NFDMP in PBS-T (1:3000) for 1 h in a humidified incubator (21 °C, 80% RH). Assay plates were washed 6x with PBS-T (100 μL/well) followed by the addition of goat anti-rabbit-HRP IgG (Jackson ImmunoResearch, West Grove, PA) diluted to 1:5000 in 5% NFDMP in PBS-T and incubated for 1 h in a humidified incubator (21 °C, 80% RH). After incubation, assay plates were washed 6x with PBS-T (100 μL/well). Luminescence was developed using 20 μL/well of luminescent substrate (Pierce West Pico PLUS, ThermoFisher, Waltham, MA) for 15 min at RT Ultrasensitive luminescence was read on an Envision plate reader at 0.1 s per well.
HBGA blocking assay
Maxisorp 384-well assay plates (Thermo Fisher, Waltham, MA) were coated with human saliva (Precision for Medicine, Frederick, MD) diluted 1:500 in Dulbecco’s phosphate-buffered saline (DPBS). Assay plates were centrifuged at 1500 × g for 1 min and incubated overnight at 4 °C in a humidity chamber. Ten, two-fold serial dilutions of mouse sera were prepared in 5% non-fat dry milk powder in phosphate-buffered saline with Tween-20 (NFDMP-PBS-T) in 384-well masterblocks (Greiner Bio-One, Monroe, NC) with a starting dilution of 1:10. VLPs prepared at 1 µg/mL in 5% NFDMP-PBS-T were added in a 1:1 dilution to the mouse sera serial dilutions. Masterblocks were sealed, centrifuged at 1500 × g for 1 min and stored overnight at 4 °C in a humidity chamber. Assay plates were rinsed 6x with PBS-T and blocked with 5% NFDMP in PBS-T for 30 min in a humidified incubator (21 °C, 80% RH). VLP-mouse sera mixtures were transferred to assay plates and incubated for 2 h in a humidified incubator (21 °C, 80% RH). Assay plates were washed 6x with PBS-T. VLP was detected by incubation with GII.3-specific polyclonal rabbit serum diluted with 5% NFDMP in PBS-T (1:3000) for 1 h in a humidified incubator (21 °C, 80% RH). Assay plates were washed 6x with PBS-T. Goat anti-rabbit-HRP (Fc)-IgG (1:5000 in 5% NFDMP in PBS-T) (Jackson ImmunoResearch, West Grove, PA) was added to assay plates and incubated for 1 h in a humidified incubator (21 °C, 80% RH). Assay plates were washed 6x with PBS-T. Luminescent signal was developed for 15 min at RT with luminescent substrate (Pierce West Pico PLUS, ThermoFisher, Waltham, MA). BT50 values were defined as the titer at which luminescence readings were 50% of the positive control. A value of 10 was assigned to samples with a BT50 less than the starting dilution of 20. For all experiments, an anti-GII.3 mouse serum sample was included as a blocking control, and plates were rejected if the BT50 for the blocking control was greater than one dilution above or below the known BT50.
Serum IgG and human GII.3 monoclonal antibody ELISA binding assays
Maxisorp 384-well plates (Thermo Fisher, Waltham, MA) were coated with 50 ng/well of VLP diluted in Dulbecco’s phosphate-buffered saline (DPBS). Plates were sealed, centrifuged at 1500 × g for 1 min and incubated overnight at 4 °C in a humidity chamber. Human IgG1 monoclonal antibodies, discovered internally by panning against a human Fab phage display library with norovirus VLPs as antigens, or mouse study serum samples, were diluted in DPBS to a starting concentration of 200 μg/mL (antibody) or 1:50 (serum) and subsequently diluted in masterblocks in a four-fold, ten-point series in 3% non-fat dry milk powder (NFDMP) in PBS containing 0.1% tween-20 (PBS-T). Dilution masterblocks were sealed, centrifuged at 1500 × g for 1 min and stored overnight at 4 °C in a humidity chamber. Assay plates were washed 6x with 1X PBS-T (100 μL/well) and blocked with 3% NFDMP in PBS-T (80 μL) for 30 min in a humidified incubator (21 °C, 80% RH). Monoclonal antibodies or serum were added to 384-well assay plates and incubated for 2 h in a humidified incubator to allow for VLP binding (21 °C, 80% RH). Following incubation, the assay plates were washed 6x with PBS-T (100 μL/well). VLP-bound antibodies were detected with HRP-goat anti-human IgG, Fcγ (min X) (Jackson ImmunoResearch, West Grove, PA, Catalog #109-035-098) or goat anti-mouse IgG (Fc), HRP conjugated (ThermoFisher Catalog #A16084) in 3% NFDMP in PBS-T (1:10,000) for 1 h in a humidified incubator (21 °C, 80% RH). Assay plates were washed 6x with PBS-T (100 μL/well) and developed with luminescent substrate (Pierce West Pico PLUS, ThermoFisher, Waltham, MA) for 15 min at RT. Ultrasensitive luminescence was read on an Envision plate reader (PerkinElmer, Waltham, MA) at 0.1 s per well. Interpolated serum antibody endpoint titers were calculated as the highest dilution where the relative light unit (RLU) signal is above a 50,000 RLU cutoff. Samples where no dilution crossed the threshold were given a placeholder titer of “25”. For samples above the threshold at the highest dilution tested (1:13107200), the value 13107200 was used for the titer. Samples where a well value was the same as 50,000 RLU were given the interpolated titer value at that exact dilution. If a sample crossed the 50000 RLU threshold between two dilutions, the titer at the crossing point was calculated by connecting the nearest points above and below the threshold, solving for the dilution at which this line crossed the threshold.
mRNA/LNP generation
VP1-expressing mRNAs were generated by Trilink Biotechnologies (San Diego, CA) with N1-methyl-pseudouridine triphosphate modification and clean-cap. LNP-encapsulating mRNA was prepared by a rapid precipitation process as previously described58. The lipid components of the LNP comprised an asymmetric ionizable amino lipid, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol and poly(ethylene glycol)2000-dimyristoylglycerol (PEG2000-DMG) in a molar ratio of 58:30:10:2, respectively.
Rabbit polyclonal sera generation
The Institutional Animal Care and Use Committee at Merck & Co., Inc., Rahway, NJ, USA, approved the rabbit studies. Three female New Zealand white rabbits (approx. four months old at receipt) from Jackson Labs (Bar Harbor, ME) were housed singly and randomized into two groups. Animals were inoculated three times (two weeks between doses) intramuscularly in the quadriceps bilaterally. Inoculations consisted of 50 μg of appropriate VLP and 225 μg of amorphous aluminum hydroxyphosphate sulfate (AAHS) in 250 μL total (remaining volume was phosphate-buffered saline), administered once in each quadriceps at each time point. Blood draws were performed at weeks 1, 3, 5, and 6 after the first inoculation. All blood draws were from the ear artery, and the final was a terminal bleed. For all survival blood draws, animals were dosed with acepromazine (1–3 mg/kg subcutaneously) and approximately 2 mL of blood was drawn from the ear artery. For terminal bleeds, animals were administered ketamine (100 mg) and xylazine (20 mg) intramuscularly, then were bled from the ear artery until no more blood could be acquired. A cardiac puncture was performed for the remaining blood, and then euthanasia was performed by administration of Euthasol intravenously. Euthanasia was confirmed by cutting the diaphragm to cause pneumothorax. All blood was spun at 4000 ×g for 5 min to separate serum, which was transferred to new tubes.
Mouse immunization studies
The Institutional Animal Care and Use Committee at Merck & Co., Inc., Rahway, NJ, USA, approved the mouse studies. BALB/c mice aged 6–8 weeks were obtained from Charles River Laboratory (Malvern, PA). Animals were housed in a company animal facility, in accordance with the Guide for the Care and Use of Laboratory Animals, and the facility is credentialed by the Association for Assessment and Accreditation of Laboratory Animal Care59. Seven groups of mice (N = 8/group) were immunized at weeks 0 and 4 with intramuscular injections of mRNAs in a lipid nanoparticle formulation. Mice were injected with 100 μL total volume per vaccination with 50 μL injected per thigh. Two weeks after the first immunization, blood was drawn via the tail vein. Specifically, a small cut was made on the tail vein with a scalpel blade (Bard-Parker, Size 15, Fisher Catalog #0268880) and around 100–150 μL blood was collected into serum separator tubes (Sarstedt AG & Co. KG Microvette 500 Z-Gel Catalog #20.1344). Two weeks after the second immunization, all mice were euthanized via CO2 exposure. Blood collection was performed via cardiac puncture using a 1 mL syringe with a 25G needle (BD Catalog #309626). Blood was transferred into serum separator tubes. Euthanasia was confirmed by cervical dislocation. For VLP immunogen studies, nine groups of mice (N = 10 /group) were immunized with 2 μg or 0.2 μg of either GII.3 or GII.3-DS1 VLPs with and without amorphous aluminum hydroxyphosphate sulfate (AAHS) at weeks 0 and 4. Mice were injected intramuscularly with 100 μL total volume, 50 μL in each thigh. Blood was drawn at weeks 2, 6, and 10.
Statistical analysis
Statistical analyses were completed using GraphPad Prism version 10.2.2. Data were tested for normality using a Shapiro–Wilk test. If the normality of both groups is satisfied, then statistical significance was determined by an unpaired two-sample t-test with Welch’s correction. If normality is not satisfied, statistical significance was determined using a Mann–Whitney U-test.
Edman degradation sequencing
N-terminal Edman degradation sequencing was outsourced to Evotec (Princeton, NJ). Purified GII.3 VLPs were shipped at 4 °C to Evotec. The N-termini of the full-length VP1 protein and 28 kDa cleavage product were sequenced to determine the site of VP1 cleavage within the cell during expression.

Comments (0)