Materials
Cationic lipid SM-102, 1,2-distearoyl-sn-glycero-3-PC (DSPC) and cholesterol were purchased from Rhawn (China). Polyethylene glycoldimyristoyl glycerol (PEG2000-DMG) was brought from Shanghai Macklin Biochemical Technology Co., Ltd. Tetrahydrofuran (THF), Bull serum albumin (BSA), Tween-20, and trehalose were purchased from Sigma-Aldrich. Trypsin-EDTA (0.25%, w/v), fetal bovine serum (FBS), and 1% penicillin/streptomycin were bought from Gibco (CA, USA). EZ CapTM Firefly luciferase mRNA was purchased from Apexbio. Firefly Glo Luciferase Reporter Gene Assay Kit was obtained from Yeasen (Shanghai, China). Phosphate-buffered saline (PBS), DMEM medium were obtained from Hyclone Lab (UT, USA). RNase-free water and TPCK-trypsin were purchased from Thermo Fisher Scientific (UK). Trehalose Assay Kit was purchased from Nuominkeda (Wuhan, China) Biotechnology Co., Ltd. RiboGreen assay kit was purchased from Thermo Fisher. CCK8, malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) assay kits were purchased from Elabscience. RIPA buffer was purchased from Solarbio (Beijing, China). Reactive Oxygen Species (ROS) detection kit, Nuclear and Cytoplasmic Protein Extraction Kit, and Annexin V-FITC/PI apoptosis detection kit were obtained from Beyotime. Goat Anti-Rabbit IgG H&L polyclonal antibody FITC (PTB96441) and Anti-NFE2L2 polyclonal antibody were purchased from Antibody System (France).
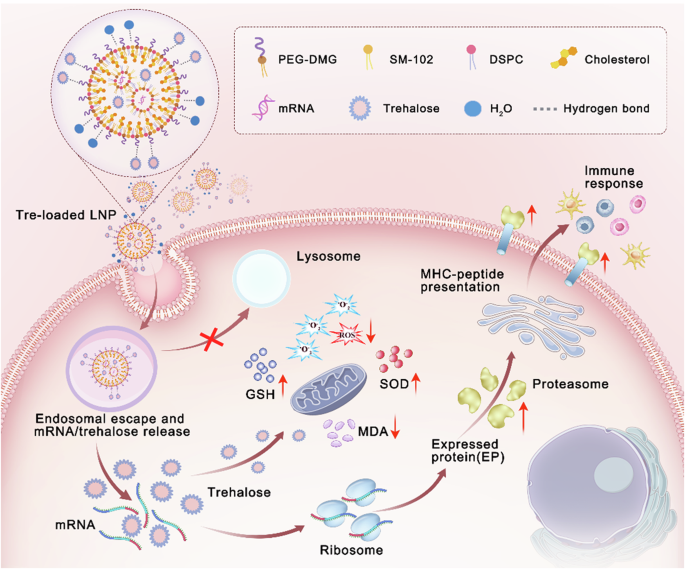
Preparation and characterization of trehalose-loaded LNP (TL-LNP) in aqueous form
mRNA-loaded LNPs in aqueous form were synthesized using a one-step nanoprecipitation and solvent evaporation method. An organic phase was prepared by dissolving SM-102, DSPC, cholesterol, and PEG2000-DMG in a molar ratio of 3.5:2.7:7:1 in THF/methanol (v/v = 6/1) mixed solvent. An aqueous phase of RNase-free water containing mRNA encoding luciferase was employed. The organic phase containing dissolved lipids is mixed with the aqueous phase containing mRNA at a volume ratio of 1:3, with an N/P ratio of SM-102 to mRNA is 6:1. The mixture is continuously stirred under a fume hood, allowing organic solvents to evaporate thoroughly, resulting in the formation of mRNA-loaded LNP formulation. For the trehalose-loaded LNP, abbreviated as TL-LNP, four lipids were dissolved in the THF/methanol mixed organic solvent according to above mentioned proportions and mixed with the mRNA aqueous solution, which contains 20% of trehalose (w/v). The trehalose-mixed LNP (abbreviated as TM-LNP) was prepared by directly adding trehalose to the obtained LNP suspension to achieve a final trehalose concentration of 20% (w/v), equivalent to 200 mg mL−1. In parallel control experiments, sucrose-formulated LNPs were prepared using identical methodologies by substituting trehalose with sucrose as the lyoprotectant: sucrose-mixed LNP (SM-LNP) through post-synthesis addition, and sucrose-loaded LNP (SL-LNP) via co-formulation with 20% sucrose (w/v) in the aqueous phase. All formulations were prepared using RNase-free water (pH 6.2 ± 0.2). The size distribution and zeta potential of all these samples were tested by DLS (Malvern, UK). Parallel formulations prepared in PBS (phosphate-buffered saline, pH = 7.4) served as controls to isolate buffer-specific effects. The morphology of the mRNA-loaded LNP without trehalose was observed by TEM. The encapsulation efficiency of mRNA was determined using the RiboGreen assay kit. Samples were diluted to an appropriate concentration with 1× TE buffer, mixed with the RiboGreen detection solution in equal proportions, and the fluorescence intensity was measured using a fluorescence spectrophotometer to calculate the concentration of free mRNA in the samples. Then, the samples were diluted with 2% Triton-TE buffer to disrupt the LNP structure and release all the mRNA. The samples were again mixed with the RiboGreen detection solution in equal proportions, and the fluorescence intensity was measured to determine the total mRNA concentration. The encapsulation efficiency of the mRNA was calculated using the following formula:Encapsulation Efficiency (%) = 1 - (Cfree / Ctotal) × 100%.
The encapsulation efficiency of trehalose within TL-LNP was assessed utilizing an ultrafiltration centrifugation technique. Specifically, TL-LNP samples were subjected to centrifugation at 5000 rpm for 2 h using ultrafiltration centrifugal tubes with a molecular weight cutoff (MWCO) of 3000 to remove the free trehalose. Then the samples were treated with 2% Triton-TE buffer to release all the encapsulated trehalose. The encapsulated trehalose was subsequently quantified via a trehalose assay kit and the encapsulation efficiency of trehalose was calculated according to the following formula: Encapsulation Efficiency (%) = (Cloaded / Ctotal) × 100%.
Cell culture
Human embryonic kidney (HEK293T) cells were obtained from the Procell Life Science & Technology Co., Ltd (Wuhan, China). Cells were cultured in high-glucose DMEM medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% (v/v) penicillin/streptomycin (Gibco) in a humidified atmosphere with 5% CO2 at 37 °C.
In vitro transfection
For the luciferase assay, HEK293T cells were seeded in 96-well plate at 5 × 104/well density 48 h before transfection to reach 60 ~ 80% confluence. Following the removal of growth medium, 190 µL of fresh serum-free DMEM medium or completed DMEM medium was added per well, and 10 µL of LNP containing 100 ng mRNA was added. After 4 h of incubation, the transfection medium was replaced with growth medium, which was supplemented with 10% FBS and 1% (v/v) penicillin/streptomycin. The luciferase activity in 50 µL medium from transfected cells was assayed with 50 µL luciferase substrate after 24 h using a GloMax® Microplate Reader (Promega), and its activity was expressed as relative light units (RLU). Similarly, the transfection efficiencies of various formulations modified with trehalose or sucrose were also evaluated.
Animals
9-12-week-old male BALB/c mice used for animal experiments were purchased from Guangdong Medical Laboratory Animal Center. The mice were maintained under specific pathogen-free conditions at the animal facility in Shen Zhen University Laboratory Animal Center. All animal experiments were performed in strict accordance with the Regulations for the Care and Use of Laboratory Animals and Guideline for Ethical Review of Animals.
In vivo bioluminescence imaging
Mice were placed into groups of n = 5 and housed in a fully acclimatized room with free access to food and water. Animals were given an adaption time of at least 7 days before experiments. Mice were injected intramuscularly (IM) in both hind leg quadriceps muscles with freshly prepared LNP, TM-LNP, TL-LNP, SM-LNP, and SL-LNP (5 μg of fLuc mRNA per leg). After 48 h, the mice were injected intraperitoneally with 100 μL of D-Luciferin substrate and allowed to rest for 15 min. Mice were anesthetized in the induction chamber with isoflurane at a flow rate of 3 L min−1 using the XGI-8 gas anesthesia system. Once anesthesia was successfully achieved, the mice were transferred to the imaging chamber and imaged on an IVIS® Spectrum In Vivo Imaging System, with isoflurane administered at a flow rate of 1 L min−1. A signal from each injection site was quantified using Molecular Imaging Software and expressed as RLU. At the end of the experiment, mice were euthanized by intraperitoneal injection of sodium pentobarbital at a dose of 300 mg kg−1.
Preparation and characterization of freeze-dried mRNA-LNP formulations
Freshly prepared TM-LNP and TL-LNP, each containing 16.7 μg mL−1 of mRNA, were stored at −80 °C or −20 °C for 24 h, followed by freeze-drying for an additional 24 h to obtain the freeze-dried samples, referred to as FDTM-LNP and FDTL-LNP, respectively. LNP without trehalose was also freeze-dried to serve as the control group. The lyophilized samples were then rehydrated and subjected to DLS analysis to evaluate particle size and zeta potential. The morphology of rehydrated FDTL-LNP was further examined using TEM. The encapsulation efficiencies of mRNA in FDTM-LNP and FDTL-LNP were also tested using the same method as mentioned above. After freeze-drying for 24 h, the resulting FDTM-LNP and FDTL-LNP samples were immediately rehydrated with 2% Triton-TE buffer to disrupt the LNP structures and release all the mRNA. Samples were then incubated at 95 °C for 1 min. A 2% agarose gel was prepared with 1× TAE and ethidium bromide (0.5 μg mL−1), and 200 ng of mRNA per well was loaded for electrophoresis at 80 V for 15 min. Then the gel was stained in EB solution (0.5 μg mL−1) for 30 min, and the RNA integrity was visualized using a gel imaging system, with pure mRNA and freshly prepared LNP serving as control groups.
Stability of freeze-dried mRNA-LNP formulations at refrigerated temperature
The obtained FDTM-LNP and FDTL-LNP samples according to the protocol mentioned above were hermetically sealed and stored at 4 °C for varying durations like 0, 1, 2, 3, and 4 weeks. The in vitro transfection efficiencies of these samples were then conducted at predetermined time intervals, and the freshly prepared LNP, TM-LNP and TL-LNP were evaluated as controls. Meanwhile, the in vivo luciferase expression of the FDTM-LNP or the FDTL-LNP samples stored at 4 °C for 2 or 4 weeks was also analyzed. Freeze-dried sucrose-formulated LNPs (FDSM-LNP and FDSL-LNP) were evaluated in parallel. The integrity of mRNA in FDTM-LNP or FDTL-LNP samples after storage at 4 °C for 1,2, or 4 weeks was finally examined using the gel electrophoresis method.
Intracellular trehalose analysis
3 × 106 HEK293T cells were seeded in a 6-well plate and cultured for 48 h. Subsequently, the Tre-mixed LNP and Tre-loaded LNP containing 0.5 μg mL−1 of mRNA were added and incubated in a serum-free medium. After 8 h, cells were washed with PBS, lysed with RIPA buffer, and sonicated to release all intracellular trehalose. Subsequently, the absorbance of the cell homogeneity was measured at 510 nm using a UV spectrophotometer following the instructions provided in the kit protocol. The intracellular trehalose was calculated based on the following formula: The intracellular trehalose content (%) = (Intracellular trehalose/Total added trehalose) ×100%.
Apoptosis analysis
3 × 106 HEK293T cells were seeded in 6-well plate for 48 h. Cells were incubated with various samples, including LNP, TM-LNP, or TL-LNP, each containing 0.5 μg mL−1 of mRNA in serum-free medium for 4 h. Following this, the medium was replaced with complete medium, and the cells were incubated for an additional 24 h. The cells were collected, stained with Annexin V and PI, and analyzed using a flow cytometer.
Cell viability
5 × 104 HEK293T cells were seeded in a 96-well plate. After 48 h, cells were treated with various formulations including LNP, TM-LNP, or TL-LNP at 0.5 μg mL−1 of mRNA in serum-free medium for 8 h. Then the medium was replaced with complete culture medium, and cells were further incubated for 24 h. Subsequently, the cell viability of HEK293T cells was evaluated using CCK8 assay.
Intracellular reactive oxygen species (ROS) detection
3 × 106 HEK293T cells were seeded in 6-well plate for 48 h. Then the HEK293T cells were treated with LNP, TM-LNP or TL-LNP, respectively at an mRNA concentration of 0.5 μg mL−1 in serum-free medium for 4 h. Then the cells were collected and resuspended in the ROS fluorescent probe DCFH-DA solution (10 μM) to a final cell concentration of 3 × 106 cells mL−1. The cell suspension was incubated at 37 °C in a cell incubator for 10 min, with gentle inversion every 3 − 5 min to ensure thorough contact between the probe and the cells. Subsequently, the cells were washed with PBS for three times and detected using a flow cytometer.
Intracellular oxidative stress evaluation
3 × 106 HEK293T cells were seeded in 6-well plate. After 48 h, the cells were treated with various formulations, including LNP, TM-LNP, or TL-LNP, each containing 0.5 μg mL−1 of mRNA in serum-free medium. After 8 h, cells were washed, digested, and collected in 500 μL of PBS and subsequently homogenized in an ultrasonic homogenizer. The homogenate was then centrifuged at 10,000×g for 10 min at 4 °C, and the supernatant was collected and kept on ice for further analysis. The malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione (GSH) contents were assessed using respective assay kit.
In vitro transfection and intracellular ROS detection of Vitamin C-incorporated LNP
Trehalose was replaced with Vitamin C (VC) for the preparation of VC-mixed LNP and VC-loaded LNP, abbreviated as VCM-LNP. The LNP, VC-mixed LNP, and VC-loaded LNP were added to HEK293T cells at concentrations of 50 µM VC and 0.5 µg mL−1 mRNA for 4 h. After 24 h, the transfection efficiency was analyzed.
5 × 104 HEK293T cells were seeded in a 96-well plate for 48 h. Similarly, the LNP, VC-mixed LNP, and VC-loaded LNP were added to HEK293T cells at concentrations of 50 µM VC and 0.5 µg mL−1 mRNA for 4 h. Then the cells were stained with DCFH-DA for 5 min and imaged by Microplate Reader (BioTek). The intracellular mean fluorescence intensity (MFI) in each group was analyzed by Gen5.
Intracellular location of Nrf2 associated with toxic and oxidative stress
2 × 105 HEK293T cells were seeded in a confocal dish for 48 h. After treatment with LNP, TM-LNP, or TL-LNP at 0.5 μg mL−1 of mRNA in serum-free medium for 12 h. Then the cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS for 15 min, and blocked with 2% BSA in PBS for 1 h at room temperature. The samples were then incubated overnight at 4 °C with Anti-NFE2L2 polyclonal antibody. After PBS washing, the cells were incubated with Anti-Rabbit IgG H&L polyclonal antibody FITC for 1 h at room temperature, followed by PBS washing. Finally, all samples were stained with DAPI and observed under a confocal microscope.
Western blotting analysis of vaccine-associated changes in Nrf2
4 × 105 HEK293T cells were seeded in a 6-well plate for 48 h. Then cells were incubated with LNP, TM-LNP, or TL-LNP at 0.5 μg mL−1 of mRNA in serum-free medium for 12 h. Cells were washed with PBS, then the nuclei and cytoplasm were separated using Nuclear and Cytoplasmic Protein Extraction Kit. Proteins were denatured by heating at 90 °C for 10–15 min. Samples were subjected to sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE) gels and transferred to polyvinylidene difluoride membranes. After blocking with 5% skimmed milk and washed with TBST, membranes were incubated with the primary antibodies and species-specific secondary antibodies. Finally, protein bands were detected by chemiluminescent analyzer using the ECL kit.

Comments (0)