
Remember me
This was a Phase 1a, single-center, double-blinded, ascending single and multiple dose study to assess the safety, tolerability, and PK of oral NNI-362 in healthy aged subjects. The study was conducted from July 2019 through November 2021, with a final study report submitted to FDA in February 2024. The study consisted of seven cohorts (A–G) of eight subjects each (six active and two placebo). Cohorts A–D were single ascending dose (SAD), with Cohort D being in a fed state and all others being in a fasted state, and Cohorts E–G were 10-day multiple ascending dose (MAD) of NNI-362 in 1% w/v methylcellulose in sterile water oral suspension. Cohorts A–D each included two Japanese subjects to compare Japanese to Caucasian/non-Japanese individuals in the NNI-362 active subjects. The study protocol was approved by the Institutional Review Board and was designed, executed, and reported according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines for Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki.
At the conclusion of each cohort dosing, the monitoring committee (i.e., the principal clinical investigator, the clinical monitor, and the sponsor) reviewed the collected data, which included PK, adverse events (AEs), changes in the clinical laboratory test assessments (hematology, coagulation, serum chemistry, and urinalysis), vital sign measurements, 12-lead electrocardiogram (ECG) results, physical examination findings, and number of subjects with suicidality changes from baseline. Based on the results of this review, the committee signed off, and the next cohort (see Table 1 for dose-limiting criteria) proceeded to dosing.
Table 1 Dose-limiting decision tableThis Phase 1a trial of new chemical entity NNI-362 in liquid formulation utilized a healthy aged population for the first-in-human trial to establish safety and PK. In Cohorts A–D, a Japanese subgroup was used for the purpose of determining potential PK differences (Japanese to all others, 1:3 ratio). The control (placebo) group contained the liquid formulation in the absence of therapeutic. The controls were at a 1:3 ratio to drug groups, as was consistent with other safety trials. For Cohorts A–D, two subjects were dosed prior to the remaining subjects as a sentinel safety measure.
Two amendments changed the original dosing scheme for Cohorts D–G (Protocol Amendments 2 and 3). The planned initial study design was for Cohort D subjects to also receive 60 mg, but in a fed state to evaluate the effect of food on bioavailability (Protocol Amendment 2). However, it was determined that the planned dose of 60 mg for Cohort D was too low to evaluate the effect of food on NNI-362 bioavailability after oral administration, and the dose for Cohort D was changed to a single dose of 120 mg. Following completion of Cohort E showing lack of absorption, Cohorts F and G were changed to SAD plus 7-day MAD using medium-chain triglycerides (MCTs), Kolliphor EL, and beeswax (95/3/2% w/w) lipid-based oral suspension (Protocol Amendment 3). Cohorts F and G consisted of a single dose followed by a minimum waiting period of 5 days and then 7 days of daily dosing.
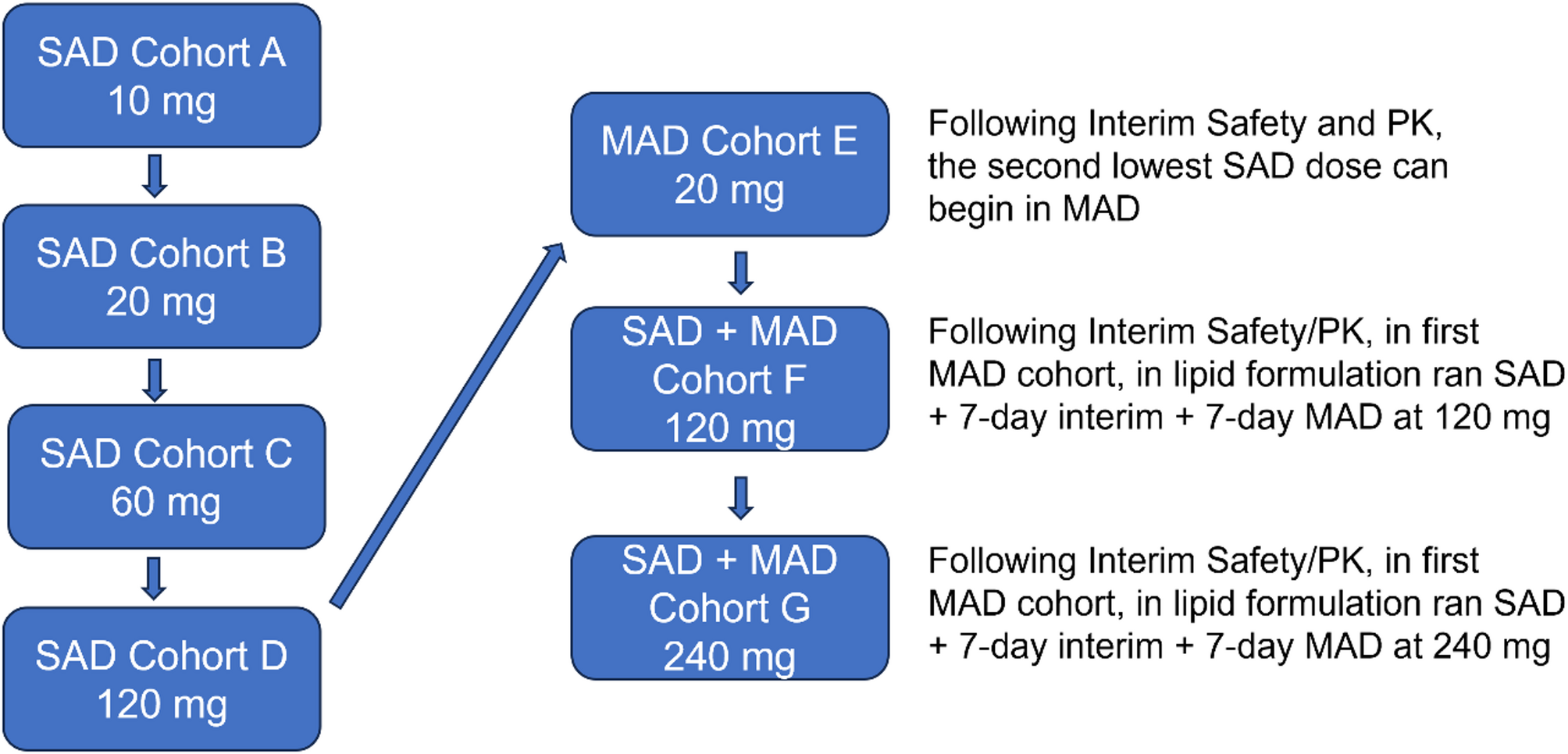
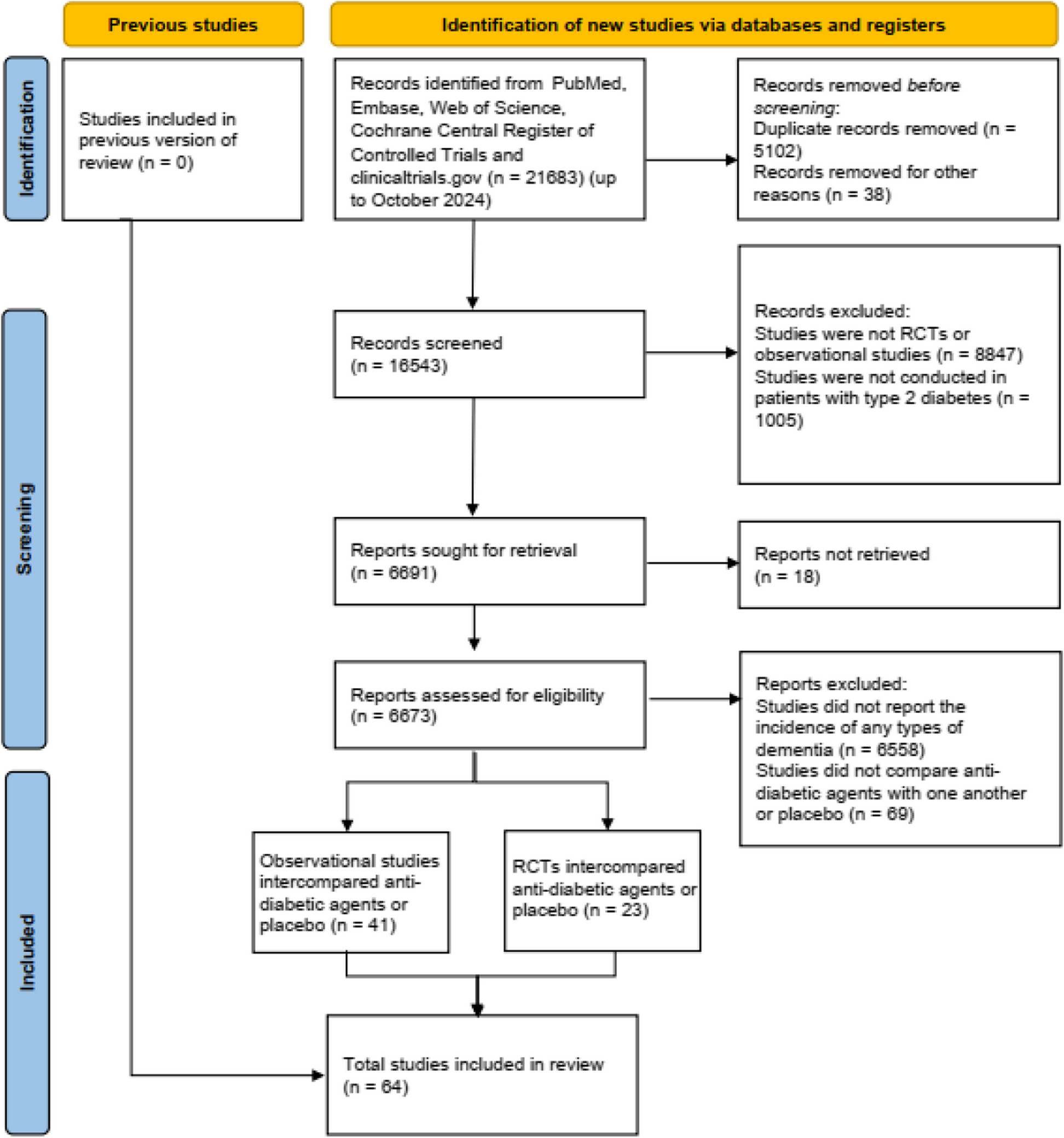
A schematic of the amended study design is included in Fig. 1 below.
Fig. 1
Study design. A schematic of the SAD and MAD dose-escalation study design. This first-in-human study of NNI-362 uses placebo control to assess the safety and tolerability in both SAD and MAD studies. MAD, multiple ascending dose; PK, pharmacokinetics; SAD, single ascending dose
The starting dose selected was based on nonclinical studies. Following single-dose administration of 10, 20, or 60 mg NNI-362, there were no safety or tolerability concerns at any of these dose levels. Following single doses of 10 and 20 mg, little to no NNI-362 was detectable in the plasma of any subject (lower limit of quantitation [LLOQ] = 25.0 pg/mL). Following a single dose of 60 mg, three of six subjects had detectable plasma levels of NNI-362, ranging from 25.2 − 69.0 pg/mL. The average observed maximum concentration (Cmax) for the three subjects was 49.8 pg/mL with a time of observed maximum concentration (Tmax) of 6 h for two of the subjects and 24 h for one subject. Due to inconsistency in PK of NNI-362 administered in the aqueous liquid formulation in Cohorts A–E, Cohorts F and G were revised from MAD to SAD/MAD cohorts to evaluate the lipid liquid formulation (Protocol Amendment 3). Subjects were enrolled under Protocol Amendment 1.
Selection of study populationThe population of healthy aged participants being of 50–72 years of age was chosen according to the National Institute on Aging statement that an aged person may start at 50 years of age. The 72-year-old cutoff aimed to ensure the ability to find healthy individuals according to the inclusion and exclusion criteria listed in Sect. 2.2.1. The study also included an ethno-bridging component to compare Japanese to Caucasian/non-Japanese individuals for Cohorts A–D. A single site was chosen for this Phase 1a trial, the California Clinical Trials Medical Group in Glendale, CA, which provided for a diverse population in terms of demographics.
Inclusion criteriaSubjects previously dosed with NNI-362 from SAD Cohorts A–D may participate in Cohorts F or G following rescreening.
Healthy aged volunteers of either sex between the ages of 50–72, inclusive. Subjects must be in reasonably good health as determined by the investigator based on medical history, vital sign measurements, physical examination, screening laboratory results, and ECG. Normal age-related findings as well as well-controlled, chronic, and stable medical conditions (e.g., hypertension, osteoarthritis, non-insulin-dependent diabetes mellitus, osteoporosis, gout, Paget’s disease, and hypothyroidism) will not be exclusionary if they are not expected to compromise subject safety, study conduct, or study objectives.
Noninteracting medications for stable, allowable medical conditions are permitted following review and approval by the medical monitor.
Adequate understanding of the requirements of the study, provision of written informed consent, and agreement to abide by the study restrictions.
Negative urine screen for drugs of abuse within 24 h before the administration of the first dose of study drug and, in the multiple-dose study, upon readmission to the clinical unit from outpatient status.
Body mass index of between 18 and 30 kg/m2, inclusive, and a total body weight greater than 48 kg at screening.
Exclusion criteriaAlcohol consumption > 10 units per week or > 2 units per day. One unit will be defined as the amount of alcohol in a 12-ounce glass of beer, a 5-ounce glass of wine, or a 1.5-ounce serving of 80-proof spirits.
Any psychiatric condition that could jeopardize the subject’s safety or the subject’s ability to comply with the protocol.
Participation in any interventional clinical trial within the previous 90 days or 5 half-lives in which the subject received an investigational drug.
Difficulty swallowing.
Recipient of an organ transplant (solid or hematopoietic).
Febrile illness or significant infection within 96 h before administration of the study drug.
Hepatitis B surface antigen–positive or serologic evidence of infection with hepatitis C virus or human immunodeficiency virus, with the exception of hepatitis B surface antibody.
History of allergy to any component of the investigational drug formulation or placebo.
Lifetime history of suicide attempts or suicidal ideation in the last 2 years prior to screen, as determined by the Columbia-Suicide Severity Rating Scale (C-SSRS).
Women of childbearing potential, defined as premenopausal (unless the potential research subject has previously undergone hysterectomy and/or bilateral salpingo-oophorectomy).
Pregnant or breastfeeding.
Any clinically significant hematology, chemistry, coagulation, or urinalysis value at screening and Day − 1. Abnormal liver enzymes (alanine aminotransferase and/or aspartate aminotransferase > 1.5 × upper limit of normal) applies at screening and Day − 1.
Serum creatinine > upper limit of normal applies at screening and Day − 1.
Hemoglobin < 13 g/dL for males or < 11.5 g/dL for females, leukocytes < 3.0 × 103/µL, absolute neutrophil count < 1000/µL, or platelets < 150 × 103/µL applies at screening and Day − 1.
Current smoker.
Glomerular filtration rate < 50 mL/min based on Cockcroft-Gault calculation using ideal body weight at screening.
Active substance abuse.
Any significant medical illness that could compromise the interpretability of study data or affect subject safety, including but not necessarily limited to:
Chronic pulmonary disease or sleep apnea.
Clinically significant cardiac arrhythmia (either at screening or based on history).
Congestive heart failure, valvular heart disease, or ischemic heart disease.
Pulmonary hypertension.
Any disorder of the kidney or urinary tract.
Active peptic ulcer disease, gastrointestinal bleeding, inflammatory bowel disease, or chronic pancreatitis.
Liver disease (excluding Gilbert’s syndrome).
Any neurologic disorder other than chronic Bell’s palsy.
History of malignancy that has not been cured or in complete remission for at least 10 years (excluding resected nonmetastatic basal cell carcinoma).
History of seizure activity other than early childhood.
Any traumatic brain injury in adulthood.
Removal of subjects from therapy or assessmentSubjects were free to withdraw from the study at any time and for any reason. The predetermined reasons for removal of subjects from the study as well as the planned follow-up are provided in the protocol.
A subject may be terminated from the study themselves or at the discretion of the study nurse and/or principal clinical investigator for the following reasons:
AE.
Violation of the protocol.
An exclusion criteria occurs or was not immediately identified during the pre-screen.
Subject withdraws consent.
Subject does not complete full study.
TreatmentSubjects received NNI-362 or placebo, as specified in Table 2.
Subjects fasted 8 h prior to NNI-362/placebo administration. While in clinic, the subjects were fed three prepared meals available at regular times. The daily caloric intake was approximately as follows:
Kcal 2800.
Protein % 16%.
Carbohydrate % 55%.
Fat % 29%.
Saturated fat % <12%.
Salt < 6 g/day.
The prepared meals did not contain grapefruit, pomelos, or Seville oranges or foods that interfere with microsomal metabolism and must not be taken from pre-study (14 days minimum) to end of study. Subjects did not smoke or use tobacco/nicotine patches or devices. All subjects agreed to this ban on tobacco/nicotine items and grapefruit.
After the full volume was dosed, the syringe was rinsed a minimum of two times, and the subjects drank the rinsing water. To rinse the syringe, the nurse drew up approximately 4 mL of drinking water and added an additional 1 mL of air space by pulling the plunger down. The syringe was shaken for approximately 15 s, and the rinsing water was administered to the subjects. If there was still residue remaining in the syringe after two rinses, it was rinsed until the full dose was administered.
RandomizationAfter subjects provided written informed consent and were screened, a unique subject identification number was assigned and recorded. A subject was considered enrolled in the study if all of the inclusion criteria and none of the exclusion criteria were met. A subject was considered randomized once the subject had been assigned by the unblinded study personnel to receive either placebo or NNI-362 and given a randomization number. Within each cohort, subjects were randomly assigned to NNI-362 and placebo in a 3:1 ratio.
A randomization program provided by Clinical Trial Data Services (CTDS) was utilized whereby only the pharmacy manager was unblinded in order to provide placebo or intervention to the randomized subjects within each cohort. Allocation of NNI-362 or placebo took place within each cohort separately. CTDS generated the randomization list using the Statistical Analysis System (SAS). At least one set of corresponding replacement randomization numbers was provided. Each cohort consisted of eight subjects: A sentinel dose design was followed, with a dose administered to the first two subjects (1 placebo/1 NNI-362, both Caucasian/non-Japanese), and the final 6 subjects consisted of 4 non-Japanese subjects (randomized to 1 placebo and 3 NNI-362) and two Japanese subjects (active; ethno-bridging) in the SAD study (Cohorts A–D). Placebo subjects were always non-Japanese, while all Japanese subjects received NNI-362.
Selection of doses in the studyIn selecting the human starting dose for the NNI-362 first-in-human trial, it is appropriate to consider the results of both pivotal repeated-dose toxicity studies as well as the human equivalent dose and the calculated plasma protein binding (PB) factor. The no observed adverse effect level in rats in the 14-day study was 150 mg/kg/day. Using this information, the human starting dose is estimated as follows:
150 mg/kg/day × 7 (Km for 250 g rat) = 1050 mg/m2.
1050 mg/m2 ÷ 10 (safety factor) ÷ 19.8 (PB factor) = 5.3 mg/m2.
Human starting dose (mg/kg basis):
5.3 mg/m2 ÷ 37 kg/m2 = 0.143 mg/kg × 70 kg human = 10 mg.
The rat is the study’s most sensitive species. Since the no observed adverse effect level is 150 mg/kg oral in rats and 210 mg/kg (bid) oral in dogs and bioavailability is not greater in larger animals, the higher dose of ~ 2 mg/kg (120 mg) and ~ 4 mg/kg (240 mg) is required to reach the human equivalent dose to reach targeted efficacy. This first-in-human study was not aimed at reaching a maximum tolerated dose.
Due to inconsistency in PK of NNI-362 administered in the aqueous liquid formulation in Cohorts A–E, a liquid lipid formulation was utilized for Cohorts F and G.
BlindingThis Phase 1a was blinded to everyone (subjects, sponsor, principal investigator, and study staff) except the Parexel pharmacist and a nurse, who delivered the investigational products and then returned the spent oral syringes to the pharmacy. Unblinding occurred only following completion of monitoring, database lock, and sign-off by the principal investigator.
Endpoints and statisticsSafety assessmentSafety and tolerance as the primary outcome measures were determined using clinical assessments and patient responses. Safety and tolerability endpoints included monitoring and recording of AEs, changes in the clinical laboratory test assessments (hematology, coagulation, serum chemistry, and urinalysis), vital sign measurements, 12-lead ECG results, physical examination findings, and number of subjects with suicidality changes from baseline.
Vital signs included the assessment of body temperature, blood pressure, pulse rate, and respiratory rate when in a supine, relaxed position for 10 min. Orthostatic vitals always followed after supine vital signs and after subject was standing for at least 2 min. Oxygen saturation was assessed through pulse oximetry. All information was reported in the subject’s case report form. A repeat assessment was allowed at the clinical principal investigator’s discretion.
Physical examination was performed by a physician or designee to examine general appearance, eyes, ears, nose, throat, head, chest/respiratory, heart/cardiovascular, gastrointestinal, liver, musculoskeletal, skin, thyroid/neck, and lymph nodes, as well as provide a neurological/psychiatric assessment. Assessment of suicidality was performed by a trained study team member administering the C-SSRS protocol. Body weight and height were obtained by the study team.
Pharmacokinetic assessmentsBlood and urine samples for analysis of NNI-362 levels were collected from all subjects at the time points specified in the Schedule of Activities. Subjects were dosed in the fasted state (except Cohort D) and confined for intensive PK sample procurement (blood was drawn pre-dose as well as at 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h post-dose) and study monitoring. Urine was collected post-dose at 60 min, > 60 min to 4 h, > 4 h to 9 h, and > 9 h to 24 h. Urine samples were not analyzed for drug levels due to funding limitations.
A validated liquid chromatography-tandem mass spectrometry assay was used for determination of NNI-362 in human plasma. The analytical range was 25.0–25,000 pg/mL, with 0.05 mL sample volumes. Blood samples were prepared in batches by centrifugation by trained personnel and occurred within 30 min of blood collection. Centrifugation was at 1500 g (~ 2500 rpm) for 10 min at set point 4 °C. When centrifugation was complete, approximately 0.500 mL plasma were aliquoted into prelabeled cryovials using an appropriate pipette. Duplicate samples were obtained, and tubes were labeled as Aliquot 1 and Aliquot 2. Samples were frozen at − 80 °C in a sample freezer until shipment to the analytical laboratory. A minimum of 1.50 mL of blood was collected from each patient per time point, with 10 time points per patient (including pre-dose). The K3 EDTA tubes were inverted 2–3 times. The time of blood draw, fasting status upon blood collection, and all relevant data were recorded as per study operating procedure. Blood in K3 EDTA vacutainers can be stored for up to 30 min on ice prior to preparation of plasma. Cold blood samples were transferred to the plasma preparation room in closed biological containers as specified for human biological samples.
For a calibration curve to be considered acceptable, at least 75% of calibration standards must be accurate to within ± 15% of the nominal concentration (± 20% at the LLOQ). For accuracy and precision experiments, individual QC samples at low, low/mid, mid, and high concentrations were considered acceptable if they were accurate to within ± 15% of the nominal concentration (± 20% at the LLOQ). The intra- and inter-batch accuracy and precision were acceptable if they were ≤ 15% (± 20% at the LLOQ). Within a run, at least 50% of the QC samples at each concentration must have satisfied the acceptance criterion, with at least 67% of the total QC samples in a run satisfying the acceptance criterion.
PK data analysis was performed on the measurable plasma concentrations of NNI-362. The plasma concentration data were evaluated using Phoenix WinNonlin™ software (version 8.3) to perform noncompartmental analysis using the linear trapezoidal method. Nominal blood collection times were used in data analysis. The dose administered in the dose group was entered into the program as mg/subject. The following PK parameters were determined for NNI-362 plasma concentrations for each subject: observed maximal plasma concentration (Cmax), observed time to reach Cmax (Tmax), area under the plasma concentration time curve up to the last blood collection time (AUClast) and extrapolated to infinity (AUCinf), the percent of AUCinf that was extrapolated (%Extrap AUCinf), terminal elimination half-life (t1/2), and mean residence time to infinity (MRTinf). Terminal t1/2, AUCinf, and MRTinf could not be calculated in some subjects due to limited data or poor fit (r < 0.8) of the terminal phase of the plasma concentration and time curve. Extrapolation of more than 20% of the AUC may result in unreliable values for the terminal parameters.
PK profile (Cmax, Tmax, AUClast, AUCinf, t1/2, and MRTinf) following single and multiple dosing was evaluated. Significant differences in selected PK parameters between the SAD and MAD groups of Cohorts F and G were evaluated using the t-test (two-tailed, paired test; P < 0.05).
PK analysis dataset was defined by the subject not being included when there was lack of linearity of the terminal phase (r < 0.8) or fewer than three data points.
Pharmacodynamic assessmentPlasma was collected at baseline (pre-dose), Day 15 (12 h after 7-day multiple dosing), and Day 16 (24 h after 7-day multiple dosing) for multiple dose administration for measurement of p-tau181 concentrations.
The duplicate frozen plasma sample from the unused PK samples (described in Sect. 2.7.2) were analyzed (blinded to treatment) at Mayo Clinical Laboratories (Rochester, MN) from the 120 and 240 mg SAD/MAD groups (Cohorts F and G). Levels of plasma p-tau181 were determined using a Simoa™ HD Analyzer (Lot Number 503008) with an Advantage V2 kit. Calibration curves of p-tau181 ranged between 0.0 and 74.6 pg/mL. Plasma biomarkers were examined at pre-dose (baseline), Day 15 (12 h post final dose), and Day 16 (24 h post final dose). Statistical analyses examined a change from the pre-dose level of p-tau181. Data was received by NeuroNascent, the blind was broken, and all data was provided for analyses to a clinical statistician, Dr. Chengjie Xiong (Lily Inc. and Washington University in St. Louis, St. Louis, MO), for significance of change from baseline at 120 or 240 mg NNI-362 and significance from placebo change from baseline.
Changes from baseline (i.e., pre-dose) in plasma p-tau181 were further analyzed by mixed models for repeated measures, including dose (the between-subject factor), post-baseline time (the within-subject factor), and their interaction as the fixed effects, in addition to the baseline level of ptau181. An unstructured covariance matrix was assumed on the repeated measures. These analyses were implemented by SAS PROC MIXED.
Comments (0)