Cell culture
The Mycoplasma-free THP-1 cells were obtained from Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan) and maintained in a humidified incubator at 37 °C with 5% CO2 in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco) and 100 units/mL penicillin, 100 μg/mL streptomycin and 292 μg/mL L-glutamine (Gibco). The culture medium of THP-1 cells was changed every three to four days. During medium change, the supernatant was removed by centrifugation, followed by a gentle wash with 1 × GKNP buffer (contains 8.0 g NaCl, 1.0 g Dextrose, 0.4 g KCl, 0.06 g KH2PO4, 0.048 g NaHPO4, and 0.012 g phenol red dissolved in 1 L double-distilled water, pH 7.2). The cells were then centrifuged again to remove residual GKNP buffer and a fresh culture medium was added for subculturing. Cells were routinely cultured and confirmed to be free of Mycoplasma contamination throughout the experiment.
Measurement of cytokine secretion
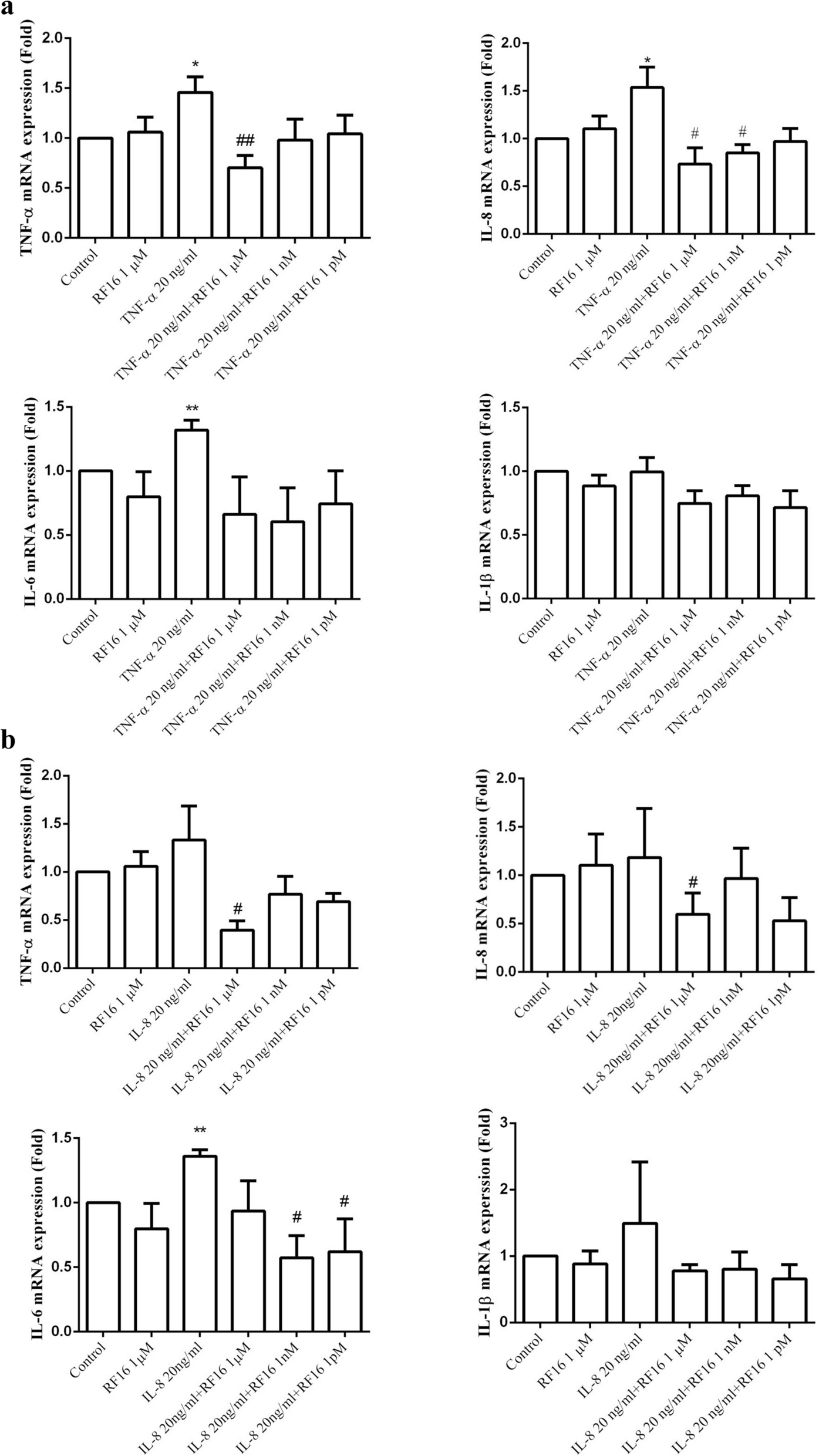
The experiment was performed as described by Chang et al. [32]. Briefly, THP-1 cells were stimulated with 40 nM PMA for 1 day to induce macrophage differentiation. After washing with 1 × Hank’s Balance Salt Solution (HBSS) buffer (contains 8.0 g NaCl, 1.0 g Dextrose, 0.4 g KCl, 0.06 g KH2PO4, 0.048 g NaHPO4, 0.2 g MgSO4, 0.14 gCaCl2·2H2O, and 0.012 g phenol red dissolved in 1 L double distilled water, pH 7.2), the cells were pretreated with RF16 peptide for 1 h followed by stimulation with cytokines (20 ng/mL) alone or in combination with RF16 peptide for 24 h. To measure the secreted levels of TNF-α, IL-6, and IL-1β, the conditioned medium was collected and analyzed using ELISA kits. All the ELISA kits were purchased from the R&D systems (Minneapolis, MN, USA), including human IL-6 DuoSet ELISA (Cat No: DY206-05), human IL-8/CXCL8 DuoSet ELISA (Cat No: DY208-05), human IL-1 beta/IL-1F2 DuoSet ELISA (Cat No: DY201-05), and human TNF-alpha DuoSet ELISA (Cat No: DY210-05).
Assays of cell viability
In order to determine the cytotoxicity of the peptide, THP-1 cells were cultured in a 96-well plate (100 μL/well) with 2 × 104 cells/well along with peptide drugs. After 24 and 48 h incubation, 8 μL of WST-1 reagent was added to each well. After 30 min of incubation, absorbance was measured at OD 450 nm. The principle of the WST-1 assay is based on the reduction of the WST-1 substrate into dark yellow formazan by cellular dehydrogenases. A higher number of viable cells results in a darker color, whereas a lower number of viable cells leads to a lighter color, indicating the potential cytotoxic effects of the tested drugs.
Detection of ROS production
THP-1 cells (4 × 104 cells/mL) were treated with 40 nM PMA and cultured in a 12-well plate. After 24 h, the cells differentiated into macrophages and adhered to the bottom of the plate. On the next day, the cells were washed with 1 × GKNP to remove PMA and then pretreated with the RF16 peptide at concentrations of 1 μM, 1 nM, and 1 pM for 1 h. Subsequently, a culture medium containing 20 ng/mL TNF-α or 20 ng/mL IL-8, along with different concentrations of RF16 (1 μM, 1 nM, 1 pM), was added and incubated for 2 h. The medium was changed to a serum-free culture medium containing a ROS detection reagent (H2DCFDA) and allowed for incubation for 10 min. Following a single wash with PBS, the fluorescence intensity of ROS was observed under a fluorescence microscope.
Phagocytosis assay of oxidized low-density lipoprotein
THP-1 cells were seeded at a density of 2 × 104 into a 24-well plate and allowed for differentiation into macrophages using 40 nM PMA. After 24 h, cells were washed once with 1 × GKNP buffer to remove residual PMA and pretreated with RF16 in serum-free culture medium for 1 h. Subsequently, a serum-free culture medium containing 50 μg/mL oxidized low-density lipoprotein (oxLDL) was prepared. In addition, 20 ng/mL of either TNF-α or IL-8, along with the respective concentrations of RF16 were added into the medium. The pretreatment medium was removed, and 300 μL of the prepared oxLDL-containing medium was added to each well. The cells were incubated for 24 h. The culture medium was discarded the following day, and the cells were washed three times with 1 × PBS. To fix the cells, 4% paraformaldehyde was added and incubated for 1 h, followed by a single rinse with distilled water (ddH₂O). The cells were incubated with 60% isopropanol (isopropanol: ddH₂O = 3:2) for 5 min and Oil Red O working solution was directly added to the cells and incubated for 2 h. After Oil Red O staining, the cells were washed 3–5 times with 1 × PBS, and imaged under a microscope. Finally, the intracellular Oil Red O stain was extracted using alcohol, and absorbance was measured.
Quantitative Polymerase Chain Reaction (qPCR) analysis
THP-1 cells were cultured in 6 cm2 plates until reaching confluence. The cells were pretreated with RF16 for 1 h, followed by treatment with cytokines (20 ng/mL) alone or combined with RF16 for 4 h. Total RNA of the cell was extracted using TRIzol reagent (Invitrogen; Carlsbad, CA, USA) in accordance with the manufacturer’s protocol. Five micrograms of RNA were used for reverse transcription with the RevertAid First Strand cDNA Synthesis Kit (Fermentas International, Burlington, ON, Canada). A quantitative real-time PCR (qRT-PCR) reaction was performed with the LabStar SYBR qPCR Kit (Bioline, London, UK) using the Roche LightCycler 480 system under the following conditions: 40 cycles of denaturation at 95 °C for 15 s, 60 °C for 20 s, and extension at 72 °C for 15 s. The oligonucleotide primers used were specific for TNF-α (forward, 5’- AGG GAC CTC TCT CTA ATC AG -3’ and reverse, 5’- TGG GAG TAG ATG AGG TAC AG -3’); IL-8 (forward, 5’- ATG ACT TCC AAG CTG GCC GTG GCT -3’ and reverse, 5’- TCT CAG CCC TCT TCA AAA ACT TCT -3’); IL-1β (forward, 5’- AAA CAG ATG AAG TGC TCC TTC -3’ and reverse, 5’- TGG AGA ACA CCA CTT GTT GCT -3’); IL-6 (forward, 5’- GCC GCC CCA CAC AGA CA -3’ and reverse, 5’- CCG TCG AGG ATG TAC CGA AT -3’) and GAPDH (forward, 5’- ACG GAT TTG GTC GTA TTG GG -3’ and reverse, 5’- TGA TTT TGG AGG GAT CTC GC -3’). Relative gene expression was calculated using the 2^(-ΔΔCT) method, and gene expression levels were normalized to GAPDH.
Western blot
Extracted protein samples were resolved by 8% SDS-PAGE gels and transferred onto PVDF membranes. The membranes were blocked with 5% non-fat milk or 5% BSA and then washed with PBS with 0.1% Tween 20 (PBS-T) or Tris-buffered saline with 0.1% Tween 20 (TBS-T). The membranes were then stained with the following primary antibodies at 4℃ overnight: p-ERK, total ERK, p-AKT, total AKT, p-P38, total P38, p-JNK, total JNK, and α-tubulin. The membranes were washed with PBST or TBST and allowed to incubate with HRP-conjugated rabbit secondary antibodies. The membranes were probed using the ECL detection reagent. Protein expression was quantified with Image J and expressed relative to each α-tubulin. The dilution of all primary antibodies was 1:1000 except P-AKT, 1:2000, while the dilution of the secondary antibody was 1:5000. The membranes were stripped with the stripping buffer and the procedures were repeated, starting from the blocking stage.
Animal model of endotoxemia
The animal experiment was performed using 8- to 10-week-old male BABL/c mice (purchased from the National Laboratory Animal Breeding and Research Center, Taipei). The mice were housed in a temperature-controlled, light-cycled facility, and the experiment was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal experiment was performed by Dr. Shih-Yi Peng and was approved by the Tzu Chi University Institutional Animal Care and Use Committee (Permit Number: 104077). To induce acute sepsis, mice were injected with 1 mL of 10 mg/kg lipopolysaccharide (LPS) via the tail vein. Mice were then separated into three groups (3 mice for each group), including sterile saline, LPS combined with saline, and LPS combined with RF16 groups. After 4 h of LPS injection, sterile saline or RF16 peptide (5 mg/kg dissolved in sterile saline) was administered via the tail vein. Mice were sacrificed 24 h later, and blood was collected. The number of white blood cells and levels of pro-inflammatory cytokine IL-6 in the serum were measured.
Determination of serum IL-6 in mice
Blood samples were collected from the mice and centrifuged at 3,000 rpm for 10 min at 4 °C to obtain serum. Serum IL-6 levels were measured using a Mouse IL-6 DuoSet ELISA (Cat No: DY406-05).
Statistical analysis
All experiments were repeated at least three times. Results were analyzed using one-way ANOVA and expressed as mean ± SEM. A P-value < 0.05 was considered statistically significant.




Comments (0)