
Remember me
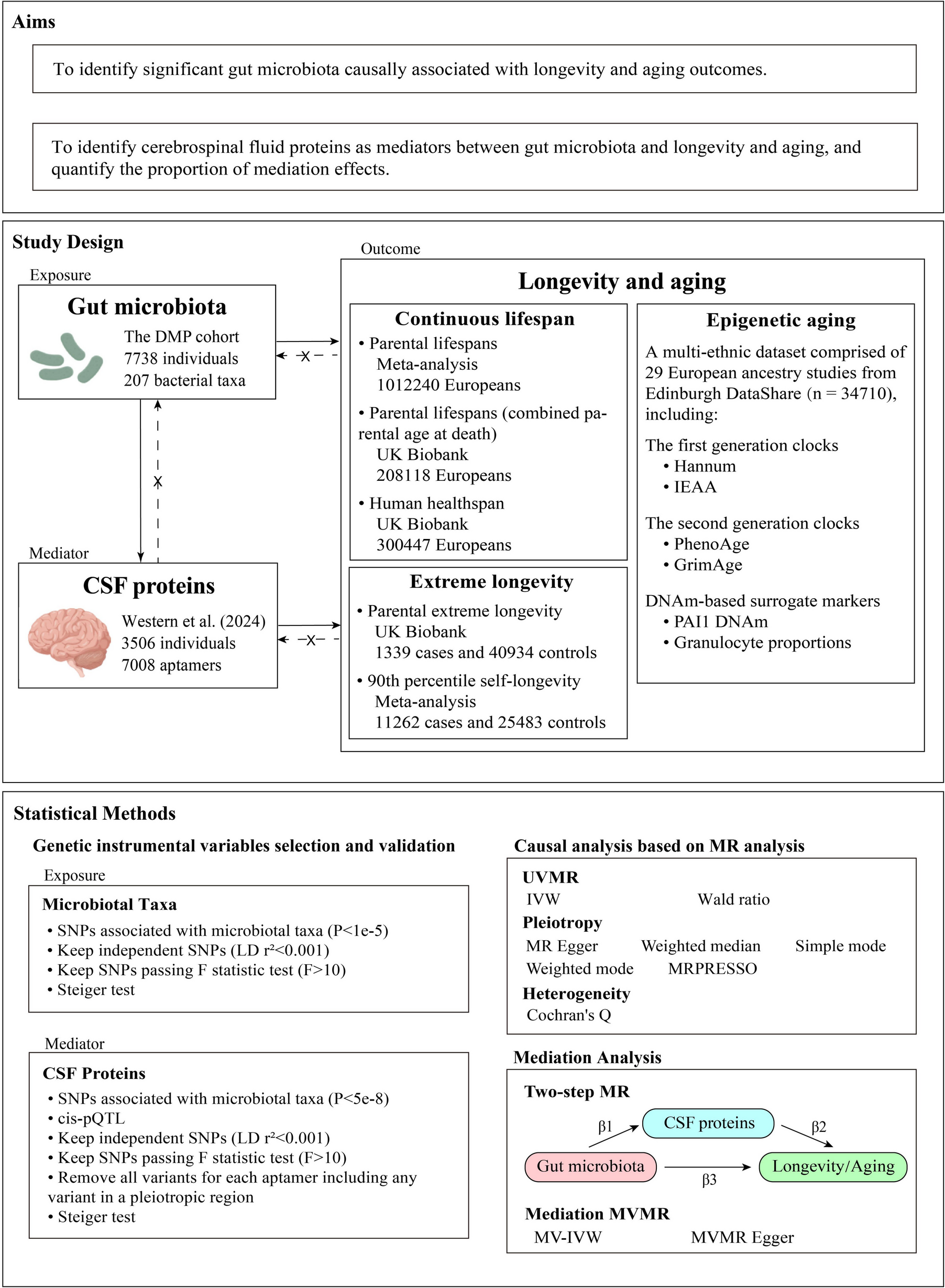



Our initial experiments were designed to serve as proof of concept for somatic mutations affecting methylation readouts. We wanted to examine a condition with both apparent methylation changes and a high number of somatic mutations, especially C>T. Our collaborators brought us new data from their previously described cancer transformation studies [30]. Briefly, YAMC cells are transformed by exposure to polarized macrophages (M φ) or 4-hydroxy-2-nonenol (4-HNE). This meant we had mutations from the exposures proper which might further be clonally expanded by the culture model — greatly increasing our ability to detect somatic mutations.
We performed high-coverage whole-genome shotgun sequencing (WGSS, average coverage 30 ×) to detect mutations and BOCS (average coverage over targets 15 ×) to detect DNA methylation. In addition to analyzing the WGSS data for variants, we processed the “unconverted” reads using the same tool we used for calling methylation from the BOCS libraries [37]. This let us generate “pseudomethylation” measurements, or the apparent methylation state of raw DNA. These together allowed us to explore the relationship between DNA methylation and somatic variants (Fig. 2). The BOCS-derived methylation level showed the expected bimodal distribution (Fig. 2A). Meanwhile, the WGSS-derived pseudomethylation exhibited a Zipfian distribution with a mode at 100%. This is consistent with most loci matching the reference genome, and the tail being created by mixed C/T reads induced by mutations. In addition, we observed a negative correlation between methylation level and variant allele frequency (Pearson r = − 0.2597). Since these data were true pairs (aliquots of DNA from the same samples), we compared the bisulfite methylation levels to both pseudomethylation and independently performed variant calls (“Methods,” Supplemental Table 1). We restricted the analysis to loci with greater than 10 × coverage in the bisulfite-tolerant alignment performed by Bismark in both the BOCS and WGSS datasets, leaving us with 145,667 loci detected in both unconverted and converted sequencing data across all samples (Fig. 2C). We further restrict some analyses of these common sites with independently called variants by intersecting the loci covered in the methylation readouts with SNVs/indels using bedtools, leaving 763 confidently measured methyl/pseudomethyl/variant triples. These loci with confident variants exhibited both a reduction in their bisulfite-determined methylation level, as well as a distribution reminiscent of the overall BOCS distributions generated by unconverted WGSS sequences (Fig. 2B). We observed that the correlation between methylation signal and “pseudomethylation” from WGSS was low (Pearson r = 0.0914) in the overall data (Fig. 2C). The correlation is further depleted when removing any loci with mutations (Pearson r = 0.0400), and greatly enriched in mutation-adjacent loci (Pearson r = 0.5750) (Fig. 2D). We then attempted to correct for the loss of methylatable cytosines occurring at the mutated loci. To do this, we divided each locus’ methylation level by its pseudomethylation level, generating an M/P ratio. Interestingly, loci across the distribution of observed methylation levels were completely explained by pseudomethylation, as exhibited by sites with M/P of 100 despite lower percentages observed in BOCS reads (Fig. 2E). While we originally hypothesized that C>T mutations would simply drive methylation estimates down, we also observe many loci with 0 M/P despite observing non-zero methylation levels (Fig. 2E). In order for this value to occur, the denominator pseudomethylation value must be near zero (implying near complete C>T conversion compared to reference) while the bisulfite-seq observes some proportion of methylated reads. We believe this effect is driven by the difference in selection strategy, since the whole-genome reads are derived from less-biased shotgun sequencing, while the bisulfite data relies upon capture probes which may bias toward methylated reads to survive capture selection, or alternatively is driven by the known bias of bisulfite-based sequencing which can enrich libraries for methylated regions [41]. This may reflect an observational bias toward unmutated sequences, due to either capture probe stereochemistry/kinetics or by methylated reads being more likely to survive the harsh bisulfite treatment. While these results were exciting for our proof of concept experiment, their extent and frequency must be understood. Even in an in vitro system including induced mutagenesis, most of the loci are unaffected by mutation readouts (Fig. 2F). However, some loci have their apparent methylation level misestimated by 50 + %. These results suggest that mutations do in fact influence methylation readouts, and it may be possible to deconvolve methylation signals with paired mutation distributions.
Fig. 2
Inducing somatic mutations in vitro reveals the influence of mutations on epigenetic readouts. Bisulfite oligonucleotide capture sequencing (BOCS) and whole-genome shotgun sequencing (WGSS) data from cancer clones (n = 6) were analyzed using Bowtie2 and Bismark. A Overall distribution of methylation (BOCS) and pseudomethylation (WGSS) at all sufficiently covered loci. B Distribution of methylation and pseudomethylation in loci with confident mutation calls. C Relationship between pseudomethylation and methylation at all loci (Pearson r = 0.0914). D Relationship between pseudomethylation and methylation at confident mutation-called loci (Pearson r = 0.5750). E Relationship between standard bisulfite and bisulfite sequencing adjusted for pseudomethylation level (methylation/pseudomethylation, M/P) at each locus. F Distribution of changes in apparent methylation (methylation — (methylation/pseudomethylation)) across all loci
Epigenetic clocks can be trained on microarray data without bisulfite conversion of DNAGiven our demonstration that it was possible for genetic information to leak into methylation readouts in sequencing data, we were curious if the changes driving pseudomethylation levels in sequencing contexts also affect methylation microarrays and, subsequently, epigenetic clocks. We hypothesized that chemical alterations to DNA other than methylation may occur during aging, other time-related processes [42], or induced during sample prep, that could be detected unintentionally by methylation arrays and contribute to age predictions. To this end, we generated two datasets from 38 samples split for parallel processing. The first dataset was processed as indicated by Illumina [13], generating apparent methylation data from bisulfite-converted DNA samples (converted), while the second was generated using the same workflow but without the addition of sodium bisulfite (unconverted). These datasets were then used to train and evaluate age-predictive models and explore their respective information content. In order to evaluate not just our bespoke dataset, but human epigenetic clocks and the properties of unconverted data at different scales, we approached this problem from multiple directions. We analyzed the age-predictive loci from our converted and unconverted clocks, and by published human clocks, to identify commonalities that may help us understand if human clocks are affected. Finally, we tested the ability of human genotyping array data to directly predict age by training “clocks” on both allele calls and raw array signal.
Since we could not locate public datasets using unconverted DNA on methylation arrays, and to leverage the paired dataset we have generated, we took a two-layered cross-validation approach. In addition to cross-validation within training data during the regression, we iteratively split our data into 75/25 train/test splits. We then generated 100 clocks for each resampling of both converted and unconverted datasets (Fig. 3) to compare how often the models can predict age at all, their relative accuracies, and how the loci chosen affect these outcomes. Significant main effects were detected for both model score (F(5, 595) = 77.58, p = 1.4 × 10−62) and model error (F(5,595) = 41.49, p = 1.2 × 10−36). We first observed performance significantly better than the dummy or permuted groups from both converted (Fig. 3A) and unconverted (Fig. 3B) models trained on the same train/test split (full significance testing in supplemental Table 2). The models trained on unconverted loci had significantly lower accuracy and higher error than the real methylation data, and training models with both converted and unconverted features did not significantly increase performance, implying redundancy in information. These findings were recapitulated even without primary feature selection (Supplementary Fig. 4) for both score (F(2,297) = 86.66, p = 3.06 × 10−29) and error (F(2,297) = 38.90, p = 2.06 × 10−15). Taken together, these results show we can generate “epigenetic” clocks even without the application of sodium bisulfite which encodes the methylation status into a discernable base difference. However, it is still undetermined if the methylation array signal detected with unconverted DNA is directly caused by DNA mutations or rather influenced by local changes to the genome and epigenome.
Fig. 3
Epigenetic clocks can be trained on mouse methylation microarray data with or without bisulfite conversion. DNA was isolated from dissected mouse hippocampi throughout the mouse lifespan (n = 38). In parallel, DNA was processed with standard bisulfite conversion (converted) or a mock conversion process lacking solely the bisulfite reagent (unconverted) for each of the 38 samples. Models were trained on converted and unconverted data. Example fits are shown for one iteration’s test set for converted (A) and unconverted (B) data. To obtain an estimate of age prediction accuracies, 75/25 train/test splits were repeated for 100 iterations to obtain distributions of prediction accuracy (C) and error (D). The distribution of observed model scores and errors were compared using one-way ANOVA and Tukey’s post hoc tests with two-tailed significance testing. Models trained on converted, unconverted, and both datasets combined were all significantly better than a dummy model predicting the mean age, as well as models trained on samples with permuted ages. The converted models were also significantly better than unconverted alone or both
Understanding the relationship between converted and unconverted epigenetic clocksThe observation that unconverted DNA could produce comparable epigenetic clocks led us to question if the clocks were detecting common signals shared between the converted and unconverted datasets, or if the underlying molecular changes were unique to each data type (Fig. 4). To explore this, we analyzed common age-predictive sites of each data type with age (Fig. 4C), the direction of predicted “age acceleration” within the same train/test split (Fig. 4B), as well as their data distribution (Fig. 4A) and genomic distribution (Fig. 4D). We observed that most of the loci selected by one data type were not age predictive in the other by multiple metrics. First, we found that sites with high mutual information (MI) in either converted or unconverted were largely unique — having a large peak at 0 MI across data types (Fig. 4C). Some loci exhibited comparable MI in both data types and are likely candidates for whatever information explains unconverted clocks leaking into epigenetic data. The sites that appeared in at least one clock from either modality were close to randomly expected overlap (representation factor 0.98, p = 0.679), again implying that the unconverted and converted models are neither biased toward nor against each other. The correlations of the commonly selected loci with age were generally positive in the converted and negative in the unconverted, but all combinations were observable (Suppl. Fig. 1). The absolute differences in apparent “methylation” and their variability were noticeably larger in the unconverted samples (Fig. 4A). Despite their seemingly unique age-predictive information, there was not an obvious difference in their biological characteristics. There was no statistical difference in their abundance across CpG islands or coding features (Fig. 4D). Despite the orthogonality of their underlying data, there was a significant positive correlation between age acceleration across modalities (Fig. 4B).
Fig. 4
Converted and unconverted DNA are predominantly age predictive due to unique signals but share genetic features and overall trends. A Three example loci that were age predictive in both data types. Lines show linear fit for unconverted (orange) and converted (blue). All common loci’s correlation to age can be seen in supplemental Fig. 1. B Comparative age acceleration on the test set of converted and unconverted clocks shows a positive association of age acceleration (Pearson r = 0.5713, p = 0.0002). C Univariate analysis shows loci that would pass primary feature selection are largely independent, but some fraction share information. D Genomic window analysis shows no over/underrepresentation of clock sites in either data type. Error bars show the 95% confidence interval for the odds ratio estimates
In summary, these findings provide evidence that (1) age-predictive DNA methylation changes are largely independent from the unconverted aging signal, but as many as 5% of loci on the mouse methylation array are age predictive with and without bisulfite conversion; (2) there is no obvious genomic region/sequence feature to enable us to exclude confounded loci a priori; and (3) the use of unconverted DNA signals as age predictors warrants further exploration due to their potential for a unique, orthogonal signal. However, these results are limited to our mouse aging dataset, and it was unclear if human epigenetic clocks might be gaining mutation-derived information. To this end, we explored the relationship between newly annotated regions of allele-specific methylation [28] and multiple human epigenetic clocks, as well as the potential of mutations alone to provide direct age prediction using public human data.
While it is known that germline mutations cast a discernable shadow onto epigenetic data by direct deamination [18], an exploration of the more subtle effects that local genomic sequence has on epigenetic modifications has been occurring outside of the epigenetic clock discussion [28]. Stefansson et al. identified quantitative trait loci (QTLs) which predicted the methylation level of proximal cytosines in an allele-specific manner (termed ASM-QTLs) in a cohort of > 7000 human blood samples. We compared the distribution of both QTLs underlying allele-skewed methylation phenomena (aQTLs), and the CpGs whose methylation these impinge upon (aCpGs) to multiple epigenetic clocks (Fig. 5C). When compared to their respective backgrounds, the chronological age predictor from Horvath (2013) [3] showed a significant overrepresentation in both aQTLs and aCpGs, implying the model could be gaining information from the overlapping QTL, as well as downstream through bona fide methylation differences they cause. PhenoAge [4], however, was neither over nor underrepresented in QTLs directly; it was enriched in loci whose methylation difference is best explained by a sequence variant. Finally, the DunedinPACE [39] clock was the most robust to these aQTLs, but still did not select against aQTLs nor aCpGs. Taken together, these findings imply that epigenetic clocks at worst are gaining direct information from genotype, and at best failing to discern between loci with real epigenetic changes and loci with other genomic alterations. Furthering this, we analyzed 256 samples from another study on allele-specific methylation patterns, which instead collected genotyping data using microarrays [27]. We again used elastic net regression to train aging clocks using both mutation calls (allele) and the fluorescent signal (signal) and found evidence they were significantly better predictors than a dummy model (Fig. 5A and B) by both score (F(2,297) = 126.94, p = 6.45 × 10−32) and error (F(2,297) = 30.08, p = 1.48 × 10−11) (see Supplemental Table 2 for full post hoc test results). However, their performance was markedly worse than epigenetic and unconverted clocks. These data imply that mutations alone do not fully explain the predictive power of either real epigenetic clocks nor our unconverted experiment.
Fig. 5
Mutations are weak age predictors alone, but downstream effects on methylation are overrepresented in human epigenetic clocks. A public dataset of human genotyping array data was used to construct clocks using trinary allele calls (AlleleCall) or fluorescence intensity directly (ArraySignal) (n = 203). Both were significantly more accurate (A) and had significantly lower MAE (B) than the dummy model despite poorer performance than converted or unconverted methylation array data. Distributions of observed scores and error were compared using a one-way ANOVA with Tukey’s post hocs for between group differences and using two-tailed significance testing. C Comparison of locations with allele-specific methylation relationship to human epigenetic clocks [28]. Loci were split into directly mutated positions (aQTL) and the CpGs they impinge upon (aCpG), as well as direct overlap and nearby (within 25 bp). Horvath’s first chronological clock was overrepresented in both aQTLs and aCpGs, while PhenoAge was enriched in only the affected CpGs. Meanwhile, loci from the longitudinal model DunedinPACE were biased against mutation-derived information but not depleted in loci downstream of mutational effects. Error bars show the 95% confidence interval of the odds ratio estimates
Comments (0)