Ethics approval
All experimental procedures were conducted at the University of Florida or the University of Pennsylvania in accordance with the relevant guidelines from the Guide for the Care and Use of Laboratory Animals. Vertebrate animal procedures were approved and followed the guidelines and regulations set by each institution’s respective Institutional Animal Care and Use Committees (protocol 202300000126 at Univ of Florida and protocol 803920 at Univ of Pennsylvania).
Animals
Adult male and female mice, 2–5 months of age, were housed in a temperature-controlled vivarium on a 12:12 h (hr) light/dark cycle with ad libitum access to food and water, except during behavioral testing. All behavioral testing occurred during the light cycle. Mice that only underwent viral injections were group housed (≤5 mice/cage) and mice with chronic implants were single housed following surgery.
Mouse lines included the following transgenic lines which were maintained on a C57BL/6J background (strain #000664; RRID:IMSR_JAX:000664, The Jackson Laboratory) and were bred in house within a University of Florida vivarium. Drd1-Cre (B6.FVB(Cg)-Tg(Drd1-cre)EY262Gsat/Mmucd, RRID:MMRRC_030989-UCD), Drd2-Cre (B6.FVB(Cg)-Tg(Drd2-cre)ER44Gsat/Mmucd, RRID:MMRRC_032108-UCD), and A2a-Cre (B6.FVB(Cg)-Tg(Adora2a-cre)KG139Gsat/Mmucd, RRID:MMRRC_036158-UCD) mice were obtained from the UC Davis Mutant Mouse Regional Resource Center (MMRRC) [30]. Drd1-tdTomato;Drd2-GFP double transgenic mice originate from that described previously [31]. Ai9 tdTomato Cre reporter mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J; RRID:IMSR_JAX:007909, [32]) were obtained from the Jackson Laboratory.
The sample size of animals in each experiment was selected based upon our prior experiences which yielded statistically robust outcomes with similar sample sizes (viz., no a priori statistical power was calculated). Data from animals with implants not appropriately targeted to their region intended (e.g., BLA, NAc, TuS), or with viral expression extending outside of those regions, were eliminated from analyses and are not reported herein. For all experiments, every attempt was made to pseudorandomize the experimental groups so that as many groups as possible would be included in any given replication. While for the behavioral experiments the experimenter was not blind due to the nature of the experiments (e.g., required pre-treatment with drugs, etc), post-processing and handling of data were both performed using semi-automated methods (see below).
Viral vectors
rgAAV.hSyn.HI.eGFP-Cre.WPRE.SV40 (Addgene #105540-AAVrg, 7 × 1012 vg/ml), AAV.Ef1α.DIO.Synaptophysin.mRuby and AAV.Ef1α.FLEX.Synaptophysin.GFP (both generous gifts from Dr Marc Fuccillo, University of Pennsylvania) [33], and AAV.hSyn.FLEx.mGFP-2A-Synaptophysin.mRuby (Addgene #71760-AAV1, 9.8 × 1012 vg/mL) were used for tracing. AAV.Ef1a.DIO.hChR2(E123T/T159C)-EYFP (Addgene #35509-AAV5, 1 × 1012 vg/ml vg/ml) was used for patch-clamp recording and for optogenetic stimulation during the optogenetic real-time place preference/avoidance task. AAV.Ef1a.DIO.EYFP (Addgene #27056-AAV5, 1 × 1012 vg/ml) was used as a control virus for the optogenetic real-time place preference/avoidance task. rgAAV.hSyn.DIO.hM4D(Gi)-mCherry (Addgene #44362-AAVrg, 1.2 × 1013 vg/ml) and rgAAV.hSyn.DIO.mCherry (50459-AAVrg, 1.8 × 1013 vg/ml) were used for chemogenetic inhibition. Both the rgAAV.hSyn.DIO.mCherry and rgAAV.hSyn.DIO.GFP (Addgene #50457-AAVrg, 7 × 1012 vg/ml) were used for retrograde tracing of BLA neurons to assess possible collateralization of inputs to ventral striatum.
Surgical procedures
For all surgical procedures, mice were anesthetized with 2–4% isoflurane (IsoFlo, Patterson Veterinary, Greeley, CO) in 1 L/min (min) O2, and head fixed in a stereotaxic apparatus while their body temperature was maintained using a 38 °C water bath heating pad. The scalp was shaved and cleaned with betadine and 70% ethanol. Following subcutaneous (s.c.) administration of Meloxicam (20 mg/kg) analgesia and local administration of the anesthetic lidocaine (lidocaine, 3 mg/kg, s.c., Patterson Veterinary) to the scalp, a small midline cranial incision was made.
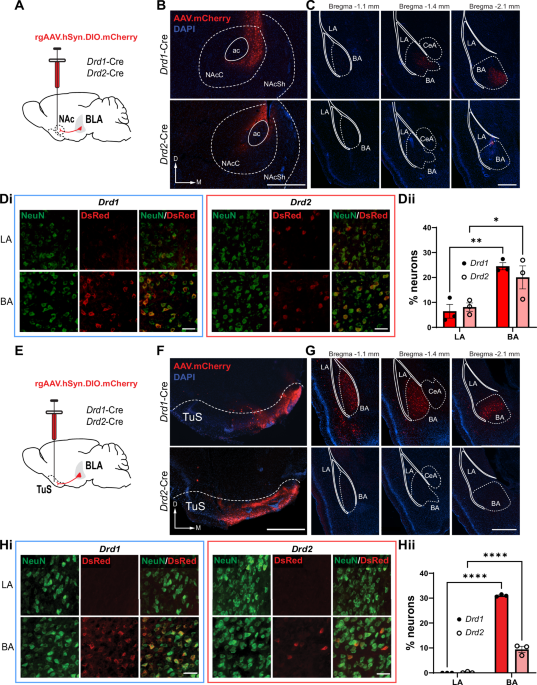
For viral injections, craniotomies were made above the target regions. A pulled glass micropipette containing the AAV was slowly inserted for injection. For TuS injections, 50 nl of viral solution was injected bilaterally at the following coordinates: anteroposterior (AP) + 1.4 mm bregma, mediolateral (ML) ± 1.2 mm lateral midline, dorsoventral (DV) −4.85 mm from the brain surface for DREADD based behaviors, or 50 nl of viral solution was delivered unilaterally for tracing experiments. For NAc injections, 100 nl of viral solution was delivered bilaterally (AP 1.5 mm, ML ± 1.0 mm, DV −3.75 mm) for behavioral experiments, or 50 nl of viral solution was delivered unilaterally for tracing experiments. For BLA injections, 100 nl of viral solution was delivered either unilaterally into the right hemisphere (AP −1.6 mm, ML + 3.25 mm, DV −4.25 mm) for Opto-RTPP/A and brain slice electrophysiology experiments, or bilaterally (AP −1.6 mm, ML ± 3.25 mm, DV −4.25 mm) for tracing experiments. All injections were performed at a rate of 2 nl/s (s), with 20–40 s intervals using a Nanoject III (Drummond Scientific). Following injection, at least 5 min went by before slowly withdrawing the pipette from the brain. Craniotomies were then sealed with dental wax and the incision was closed with wound clips.
For cannula implantation, the skull was scrubbed with 3% H2O2 and covered with a thin layer of cyanoacrylate glue (Vetbond, 3M). Bilateral craniotomies were drilled over the BLA and 26-gauge(G) guide cannulae (#C315GMN/SPC, P1 Technologies) extending 3.5 mm below pedestal were implanted at the coordinates AP −1.3 mm, ML ± 3.2 mm. Cannulae were then lowered into the brain and secured to the skull with a small amount of Vetbond followed by dental cement, and dust caps with a 3.5 mm projection wire (C315DCMN/SPC, P1 Technologies) were inserted.
For optical fiber implantation, following skull preparation for implantation as above, a craniotomy was made and drilled above the ventral striatum on the right hemisphere. Fibers (300 µM core diameter, 0.39NA, cut to 6.0 mm length) for optogenetic stimulation were lowered into the NAc (AP 1.4 mm, ML 1.0 mm, DV −3.85 mm) or the TuS (AP 1.5 mm, ML 1.2 mm, DV −4.9 mm). The fiber was secured with Vetbond followed by dental cement as described for the cannulae implantation.
Following surgery, mice were allowed to recover on a heating pad until ambulatory and were given immediate ad libitum access to food and water. Meloxicam analgesic (20 mg/kg, s.c.) was administered for at least 3 days following surgery. Mice with indwelling cranial implants were single housed and given 7–14 days after surgery to recover before being acclimated to behavioral procedures.
HistologyImmunohistochemistry
Mice were anesthetized with Fatalplus (150 mg/kg; Vortech Pharmaceutical Ltd, Dearborn, MI) and transcardially perfused with cold 0.9% NaCl (Physiological Saline), followed by cold 10% phosphate buffered formalin (#SF100-4, Thermo Fisher Scientific) for fixation. Brains were collected and further fixed and cryoprotected in a 30% sucrose/10% formalin solution for 72 hr at 4 °C. Serial 40 μm thick coronal sections were collected using a sliding microtome (Leica) and stored at 4 °C in a solution of Tris-buffered saline (TBS; 0.242% Tris base, 2.924% sodium chloride, pH = 7.4 ± 0.2) with 0.03% sodium azide.
Sections from Drd1- or Drd2-Cre mice injected with Cre-dependent retrograde mCherry AAV underwent antigen retrieval in citrate buffer (pH 6.0) for 30 mins at 80 °C. Sections from Drd1-Cre;Ai9, Drd2-Cre;Ai9, or A2a-Cre;Ai9 mice were selected to investigate recombination efficiency but did not undergo antigen retrieval. After being rinsed with TBS and diluting buffer (2% bovine serum albumin (Sigma Aldrich), 0.9% sodium chloride (Sigma Aldrich), 0.4% Triton-X 100 (Sigma Aldrich), and 1% normal goat serum (Sigma Aldrich) in TBS), samples were blocked in 20% normal donkey serum solution, then incubated in the primary antibody overnight at 4 °C. Sections were then incubated in the secondary antibody at room temperature and washed with TBS prior to slide-mounting with either DAPI Fluoromount-G® mounting medium (SouthernBiotech, catalog #0100–20). Primary antibodies included rabbit anti-DsRed (Takara Bio, catalog #632496, 1:1000), chicken anti-NeuN/FOX3 (EnCor, catalog #CPCA-FOX3, 1:1000), and mouse anti-Cre Recombinase (Millipore-Sigma, cat#MAB3120, 1:1000). Secondary antibodies included goat anti-chicken Alexa Fluor 488 (Invitrogen, cat#A11039), goat anti-rabbit Alexa Fluor 680 (Invitrogen, cat#A21109), and donkey anti-mouse Alexa-Fluor 488 (Invitrogen, cat#A21202), all at a 1:1000 dilution.
Imaging and quantification
Brain regions were identified using the mouse brain atlas [34]. Images were acquired using a Nikon Eclipse Ti2e fluorescent microscope. For quantification of the number of Drd1+ and Drd2+ NAc and TuS projecting BLA neurons, three mice of each genotype (Drd1-Cre or Drd2-Cre) received an injection in either the NAc or the TuS. From these mice, at least three BLA sections were acquired and imaged from −1.10 mm to −2.10 mm posterior to Bregma. For quantification of the number of Cre+ neurons in the BLA of Drd1-Cre;Ai9, Drd2-Cre;Ai9, or A2a-Cre;Ai9 mice, at least five BLA sections from three mice of each genotype were acquired spanning the same range. Images were acquired at 20x magnification across both hemispheres and Z-stacked every 4 µm. For quantification, regions of interest (ROIs) were drawn around the areas of interest (LA, BA). Images were preprocessed to remove background and to enhance local contrast, a rolling ball algorithm was applied to remove background, and images underwent Gaussian smoothing and Laplace sharpening. A semi-automated thresholding and counting algorithm created within NIS elements (Nikon) software was used to identify cells within selected ROIs, allowing for unbiased estimation of cell numbers. Cells were identified based on fluorescence intensity (via threshold) and diameter.
For quantification of Drd1+ and A2a + BLA to ventral striatum synaptophysin puncta within the ventral striatum, at least three sections from three mice of each genotype were acquired spanning from 1.7–0.6 mm anterior to Bregma. Images were acquired at 20x magnification for the hemisphere ipsilateral to the injection site, and Z-stacked every 0.9 µm. For quantification, ROIs were drawn around the areas of interest (TuS, NAcC, NAcSh, PCx). Images were preprocessed to remove the average background. A semi-automated thresholding and counting algorithm created within NIS elements software was used to identify fluorescent puncta within selected ROIs, allowing for unbiased estimation of the number of fluorescent puncta. Puncta were identified based on fluorescence intensity (via threshold) and diameter.
Brain slice electrophysiology
Whole-cell patch-clamp recordings were performed in ex vivo brain slices from Drd1-Cre;Ai9 or A2a-Cre:Ai9 mice, in which tdTomato expression was directed within cells expressing either DRD1 or DRD2, respectively. A Cre dependent AAV encoding for ChR2 (AAV5-Ef1a-DIO hChR2(E123T/T159C)-EYFP) was injected bilaterally into the BLA of Drd1-Cre:Ai9 or A2a-Cre:Ai9 mice, 2–3months of age. After waiting a minimum of one month to allow for ample AAV expression, acute brain slices were prepared as follows.
Mice were deeply anesthetized with intraperitoneal injection of ketamine-xylazine (200–15 mg/kg body weight) and decapitated. The cranium was dissected and the brain was immediately removed and placed in ice-cold HEPES buffered cutting solution containing (in mM): 92 N-methyl-d-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium l-ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4 and 0.5 CaCl2 (osmolality ~300 mOsm and pH ~7.4, bubbled with 95% O2 and 5% CO2). Coronal brain slice (180–200 µM) containing the OT were cut using a Leica VT 1200S vibratome. Brain slices were incubated in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2.4 CaCl2, 1.2 MgSO4, 1.4 NaH2PO4, 11 glucose, 25 NaHCO3 and 0.6 sodium L-ascorbate (osmolality ~300 mOsm and pH ~7.4, bubbled with 95% O2 and 5% CO2) for 1 hr at 31 °C and at room temperature thereafter. Slices were transferred to the recording chamber for whole-cell patch-clamp recordings and continuously perfused with oxygenated ACSF. 4-Aminopyridine (4-AP; 200 µM) was added to enhance optically evoked synaptic release in ChR2+ axonal terminals. Fluorescent DRD1-/A2A-tdTomato+ cells in the TuS were visualized with a 40X water-immersion objective under an Olympus BX51WI upright microscope equipped with epifluorescence. Electrophysiological recordings were controlled by an EPC-10 amplifier combined with Pulse Software (HEKA Electronic) and analyzed using pulse and Clampfit (Axon instruments). Whole-cell patch-clamp recordings were made in both current and voltage-clamp mode. Patch pipettes were pulled from thin-wall borosilicate glass-capillary tubing (WPI, Sarasota, FL, USA) on a Flaming/Brown puller (P-97; Sutter Instruments Co., Novato, CA, USA). The tip resistance of the electrode was 5–8 MΩ. The pipette solution contained the following (in mM): 120 K-gluconate, 10 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, 5 Mg-ATP, 0.5 Na-GTP, and 10 phosphocreatine.
To activate ChR2 in the TuS slices, blue light (pE-300ultra, CoolLED, ~25 mW) was delivered through the same 40X objective. Pharmacological reagents including tetrodotoxin (TTX) citrate (Abcam), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), d,l-2-amino-5-phosphonopentanoic acid (AP5), and 4-Aminopyridine (4-AP) (Sigma-Aldrich) were bath perfused during recording.
in vivo DREADD-based chemogenetic inhibition
For DREADD-based chemogenetic inhibition of Gi coupled inhibitory DREADD receptor (hM4Di) expressing neurons, Drd1+ and Drd2+ mice were injected with rgAAV.hSyn.DIO.hM4D(Gi)-mCherry (1.2 × 1013 vg/ml, 100 nl/hemisphere in NAc, 50 nl/hemisphere in TuS, catalog #44362-AAVrg, Addgene) or rgAAV.hSyn.DIO.mCherry (1.8 × 1013 vg/ml, 100 nl/hemisphere in NAc, 50 nl/hemisphere in TuS, catalog #50459-AAVrg, Addgene) as control. All mice were implanted 1–2 weeks later with bilateral intracranial guide cannulae (Protech International, Inc, catalog #8IC315GMNSPC, 26ga) extending 3.5 mm beyond the pedestal, for direct administration of either the DREADD ligand J60 [35] or vehicle into the BLA. Dust caps without a projection wire (Protech International, Inc, catalog #8IC315DCMNSP) were inserted immediately following surgery, and mice were given 1–2 weeks to recover.
Prior to behavior, mice underwent 2 days of handling in which the dummy cannulae were removed and replaced. On the habituation behavior day, mice received a “mock” infusion, wherein the internal cannulae (Protech International, Inc, catalog #8IC315MNSPC, 5.75 mm projection, 33ga) connected to tubing from a 1 µL Hamilton Syringe (Hamilton, catalog #86211) were inserted into the guide cannulae, and the Harvard Apparatus 22 Syringe Pump (catalog #PY2 55–2222) was turned on for 2 min to simulate the noise of the infusion. This mock infusion occurred 30 min prior to being placed in the plethysmograph for the Pavlovian fear learning behavioral paradigm. On the learning day (Day 2) of the Pavlovian fear learning paradigm, mice were once again tethered to the Hamilton syringe, but this time received an infusion of 100 nL of either 10 nM J60 or vehicle at a rate of 50 nl/min, 30 min prior to the start of the behavioral task.
Behavioral tasksOdor-shock pavlovian fear learning
We used a whole-body plethysmography chamber (Data Sciences International, St. Paul, MN) that was adapted for the infusion of a neutral odor and the administration of a mild foot shock for an odor-shock Pavlovian fear learning test, as originally developed for use in rats [36]. We constructed an air-dilution olfactometer [37, 38] and used custom code in Synapse (Tucker Davis Technologies) to control the delivery of an otherwise neutral odor, isopentyl acetate (1 torr in liquid state; Sigma Aldrich), at a flow rate of 1 L/min (20 s) which co-terminated with the presentation of a mild foot shock (0.5 mA for 1 s). Respiratory transients were detected using a Data Sciences pressure transducer, gain amplified 100 × (Cygnus Technology Inc), and digitized (0.1–20 Hz) at 300 Hz in Synapse. Positive pressure of clean room air was continuously applied to the chamber using a stable-output air pump (Tetra Whisper). Following each stimulus trial, odor-vaporized air was exhausted from the plethysmograph through an outlet at the chamber’s ceiling.
Mice were acclimated to handling in the behavioral room for two days prior to entering the plethysmograph. Mice were then acclimated to the plethysmograph by undergoing a session in which no odors or shock were delivered, but the associated sounds were present (Supplementary Fig. 5). Twenty-four hr later on the acquisition day, mice were allowed to acclimate to the plethysmograph for a 4 min period and were then presented with 10 trials of 20 s odor delivery co-terminating with an odor-paired 1 s foot shock (0.5 mA) with an inter-trial interval (ITI) of 180 s. For the unpaired fear conditioning task, the foot shock was presented pseudorandomly in the ITI (90 s after the foot shock). For the odor only control mice, the 10 trials consisted of only 20 s odor delivery without the administration of the foot shock. The shocked mice received no odor delivery during the trials but received a foot shock either at the end of the trial (trial shock group) or pseudorandomly in the ITI (ITI-shock group). Mice were then returned to their home cage. Twenty-four hr later on the retrieval day, the odor was presented for 10 trials without the foot shock for all groups receiving odor (paired, unpaired, and odor only groups). Mice who did not previously receive the odor underwent the 10 trials without odor delivery or foot shock. Mobility behavior was recorded throughout the entire fear conditioning task using two digital cameras (Microsoft, 10 Hz frame rate) and was scored in 0.4 s bins during the 19 s of odor presentation prior to shock using ezTrack [39] to identify periods of physical immobility. Respiration digitized from the pressure transducer was imported into MATLAB, and a MATLAB script was used to calculate fast-fourier transform (FFT) power spectra of the respiratory signal during odor (excluding the 1 s when the shock co-occurred) as compared to pre-odor (see Supplementary Fig. 5).
Optogenetic real-time place preference or aversion test (Opto-RTPP/A)
Mice were gently handled and acclimated to the behavior room the day prior to the opto-RTPP/A test. Prior to starting the opto-RTPP/A test, mice were gently scruffed, the dust cap was removed, and the mice were tethered to a 400 µm, 0.57NA fiber (Thorlabs, catalog #M58L01) and placed in a 15.24 × 40.64 × 27.94 cm (length × width × height) apparatus divided into three chambers. This fiber was connected to an LED (Doric, 465 nm) through a rotary joint connected to a 400 µm, 0.39NA patch cable. Mice were placed in the center of a three-chamber apparatus and allowed to explore for 30 min. An infrared video camera was placed above the chamber to record activity of the mouse in each chamber (12 Hz frame rate). When mice entered into one of the three chambers, and subsequently broke the infrared beam path, light stimulation (465 nm, 15 ms pulse width, 40 Hz) was initiated and continuously delivered until mice left the chamber and ceased breaking the infrared beams (controlled by an Arduino). At the end of the 30 min, mice were gently restrained and the tether was removed. Mice were then returned to their home cage. The mice were euthanized and perfused the same day, and brains were collected for histological verification of virus injection and optic fiber placement. Analyses were performed in ezTrack [39] to quantify the time spent in each chamber and to generate maps of physical space for illustration purposes.
Data analysis
Data were analyzed for statistical significance in GraphPad Prism. All data are reported as mean ± SEM unless otherwise noted. Specific tests used can be found in the Results sections or the figure legends. All tests met the assumptions of the test (e.g., were normally distributed, etc.) and variances between groups comparable. All t-tests were paired. When possible, experimenters handling the data were blinded to treatment conditions and all post-processing and analyses of data handled through semi-automated methods when possible.



Comments (0)