
Remember me
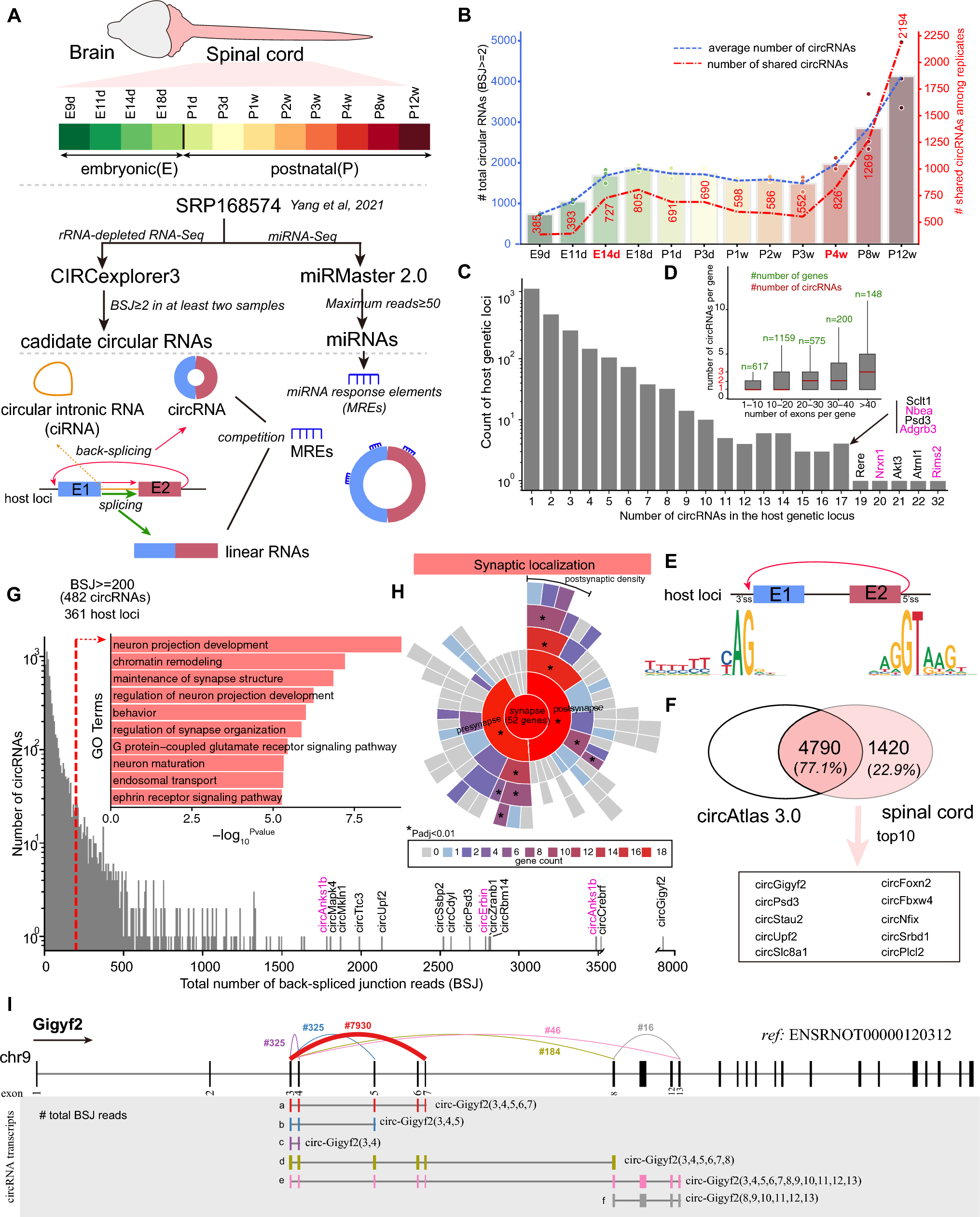


To characterize genome-wide and dynamic circular RNAs in the developing spinal cord, we analyzed our long-term time-series of rRNA-depleted RNA-Seq dataset ranging from the early embryonic stage (embryonic 9 days, E9d) to the adult (12 weeks postnatal, P12w) with a total of twelve timepoints [18] (Fig. 1A and Supplementary file 2: Table S1). We employed a robust and accurate pipeline for the identification and quantification of both circular RNAs and linear RNAs, CIRCExplore3 [19], and used the newly released rat genome (mRatBN7.2) as the reference. Both circular exonic RNAs (circRNAs) and circular intronic RNAs (ciRNAs) could be detected by the CIRCExplore3. Here we only focused on circRNAs due to a low abundance of ciRNAs (most ciRNAs with BSJ read count ≤ 2, Supplementary file 1: Fig. S1). To screen the high-confidence of circRNAs, those with at least two back-splicing junction (BSJ) reads in at least two samples from a developmental timepoint were kept for the downstream analysis. A total of 6,210 circRNA candidates generating from 2696 loci met the criterion and most were flanked by the canonical splicing motif, AG-GT (Fig. 1E). We also compared with rat circular RNAs deposited in the circAtlas database [20]. About 77.1% (4790 out of 6210) of the total circular RNAs were annotated in the circAtlas database to date based on the genomic coordinates of BSJ (Fig. 1F). Among the unannotated circRNAs, the top10 abundant circRNAs included circular transcripts generated from host loci of Gigyf2, Psd3 and Slc8a1 (Fig. 1F). Analysis of the number of circular RNAs in each time point, we found the smallest number at early embryonic stage (E9d-E11d), and increased at E14d and then small fluctuation until P3w, and gradually increased from the P4w (Fig. 1B). Most host loci generated a small number (< 10) of circRNAs but nine host loci could generate more than 17 circRNA transcripts (also known as a hotspot which contains multiple circRNAs originating from the same loci [28]), including four synapse-related genes, Rims2, Nrxn1, Nbea, and Adgrb3 (Fig. 1C). We also noticed that 248 out of 1283 loci (19.32%) with at least two circRNAs generated one major circRNA (defined with at least fivefold of the expression of the second circRNA transcript level), especially Gigyf2 (generating six circRNAs (designed as a–e) and the ‘a’ transcript was the major circRNA, Fig. 1I and Supplementary file 1: Fig. S2). Consistent with the previous report [29], the increased number of circRNAs was observed along with the increased exon number per gene (Fig. 1D). We next investigated highly abundant circRNAs in the spinal cord. Functional enrichment analysis showed that host loci generating circRNAs with a total of BSJ read count ≥ 200 were significantly enriched in terms of “neuron projection development” and “maintenance of synapse structure” (Fig. 1G). Further synaptic localization annotation and enrichment analysis showed that 52 out of 361 host loci were located at synapse and the most were located at postsynaptic density (Fig. 1H), which was consistent with a previous report in the mouse brain [11]. Taken together, these results displayed and characterized circular RNAs in the developing rat spinal cord.
Fig. 1
Genome-wide characterization of circRNAs in the developing rat spinal cord. A The schema of the analysis. B The number of the average (left y-axis) and shared circRNAs among replicates (right y-axis) in each developmental time point. C The summary of circRNA number generated by per host loci. D The summary of the relationship between circRNA number and exon number. E The motif pattern of circRNAs detected in developing spinal cord. F The overlap of circRNAs identified in developing spinal cord and circAtlas 3.0. G The high abundance of circRNAs (total BSJ read count ≥ 200) and functional enrichment of host loci. H Enrichment of synaptic location geneset for host loci generating a high abundance of circRNAs (total BSJ count ≥ 200). I Six expressed circRNAs generated from Gigyf2. Colorful link lines indicated BSJ from different circRNAs and number labelled text indicated the total BSJ count in developing spinal cord. Number in the brackets indicated included candidate exons in circRNAs
Highly abundant circRNAs in CNS but not in PNSIt is commonly acknowledged that the brain harbors a significantly higher abundance of circular RNAs (circRNAs) in comparison to other tissues, such as the liver, lung, and heart. While previous studies have delved into the analysis of circRNAs in 11 tissues (brain and various non-nervous system tissues) of developing and aging rats at different life stages (2 w, 6 w, 21 w, and 105 w) [30, 31], these investigations remain incomplete and somewhat understated due to the relatively short length of sequencing reads (~ 50 bp). Moreover, there is still a notable gap in the systematic comparison of circular RNAs between the central nervous system (consisting of the brain, spinal cord, and retina) and the peripheral nervous system (PNS, encompassing structures like the dorsal root ganglion (DRG) and sciatic nerve), as well as other diverse tissues in rats.
Fortunately, Krause et al. have recently constructed an extensive gene expression atlas encompassing four preclinical species, among which is the rat, covering up to 47 tissues (88 samples, encompassing female and male samples of the brain, spinal cord, retina, and sciatic nerve) across 12 major organ systems [32]. This endeavor was achieved using an rRNA-depleted and paired-end sequencing strategy with a read length of 75 bp (PE75), thereby providing an invaluable resource for exploring circRNAs throughout the rat's body, particularly within the nervous system. Hence, we next investigated circRNAs in rat diverse organ system using this valuable resource as well as previously independent public related datasets, including retina, spinal cord, DRG and sciatic nerve after injury [33, 34] (Fig. 2A). We first performed quality-check of samples from the rat body dataset and discarded those samples with a high percent of rRNA (> 30%) and finally kept 72 samples from 38 tissues (e.g., brain, spinal cord, sciatic nerve) with at least 50 million (M) uniquely mapped reads for the analysis (Fig. 2A and 2B, Supplementary file 3: Table S2). It showed the highest number and abundance (estimated by the total BSJ read count) of circRNAs in CNS tissues (cortex, cerebellum, medulla, spinal cord etc.), with the testes showing the next highest level compared with other tissues, including sciatic nerve (Fig. 2B). Of the different brain regions, circRNAs in cerebellum showed the most abundance (Fig. 2B), which is also consistent with the observation in mouse [10] (e.g., 3046 circRNAs in cerebellum, 2353 circRNAs in cortex using the same pipeline, Supplementary file 1: Fig. S3). We also confirmed circRNA abundance in other public datasets from brain, retina and spinal cord, and other PNS tissues (DRG and sciatic nerve). As observed, the number and abundance of circRNAs in adult retina and spinal cord were greater than that in adult PNS tissues (e.g., highly expressed circRNAs (BSJ read count ≥ 20 in at least a sample (without replicate) or two replicates)) was greater than 130, while that in PNS tissues was less than 70, Fig. 2C). Interestingly, in our analyzed datasets, we noticed that global abundance of total circRNAs in retina after ischemic stroke injury (12 h) was increased while global abundance of total circRNAs in spinal cord was obviously decreased after 3 days post hemi-transection injury (Fig. 2C). One example is a circRNA generated from the exon 4 of rat Zranb1 loci (ENSRNOT00000023257) that showed a markedly increased expression in the injured rat retina induced by glaucoma [35]. However, we found expression of most circRNAs generated from Zranb1 loci in rat spinal cord upon SCI were decreased (Supplementary file 1: Fig. S4), suggesting different expression patterns of circRNAs in injury response of distinct neural tissues. Taken together, by integrating available rat total RNA sequencing datasets and a unified analysis workflow, we showed that the highest abundance of circRNAs in CNS tissues, specifically in the cerebellum, compared with that in PNS tissues and other organs.
Fig. 2
Highly abundant circRNAs in CNS but not in PNS. A The schema of analysis workflow and was created with BioRender.com. B The number and abundance of the circRNAs in the rat body. The upper panel showed the number of circRNAs under different thresholds. The middle panel showed the ratio of circRNAs (orange) and uniquely mapped reads (blue). The bottom panel showed the top 10 abundant circRNAs. Circle colors indicate CIRCscore and sizes indicate the relative expression abundance respectively. C The circular number and abundance in rat retina, spinal cord, DRG and sciatic nerve in other studies. D The top 20 abundant circRNAs in three tissues from two species using rRNA-depleted or further with RNase R treatment. Text labelled showed host loci generating the top 5 circRNAs as well as conserved circRNAs in between mouse and rat. Link lines between rat circRNAs and mouse circRNAs indicated orthologous pairs
In addition, circRNAs generated from genetic loci of Gigyf2, Anks1b, Erbin, Ssbp2, Nfix, Cdyl, Psd3, Ttc3, Crebrf, and Zranb1, ranked the top 10 of the abundance in the rat body dataset based on the total BSJ read count (Fig. 2B). Except for a circRNA generated from a CNS-specific gene (Anks1b), others top abundant circRNAs were broadly expressed in most tissues (e.g., circ-Gigyf2). Of these, circRNAs generated from Gigyf2, Nfix, Cdyl and Zranb1 showed a higher expression than that for cognate linear transcripts with a CIRCscore > 1 (namely FPBcirc > FPBlinear, Fig. 2B) in most tissues. We noticed other two circRNAs (circ-Ssbp2 and circ-Crebrf) showed an obvious higher CIRCscore in CNS tissues than that in other tissues. Unlike in rat, the most abundance of circRNAs in mouse include circRNAs generated from host loci, Rims2, Cdr1os, Anks1b, Gigyf2, Elf2, using a total RNA-Seq dataset from mouse 14 tissues [9] (Supplementary file 1: Fig. S5). We then checked this phenomenon using another public dataset from the same study [28] and found that the most abundant circRNAs differed in tissues between rat and mouse, specifically in testis (the top 20 were shown in Fig. 2D and Supplementary file 1: Fig. S6). For example, a Zpbp-derived circRNA ranked the top in both the total RNA Sequencing and RNase R treatment datasets from rat testis, but a Bbs9-dervied circRNA ranked the top in both the total RNA Sequencing and RNase R treatment datasets from mouse testis (Fig. 2D). The well-explored circular Cdr1as (also known as ciRS-7), originating from a long non-coding RNA locus (Cdr1os), is the most abundant in human and mouse brains [5, 36]. However, we noticed that Cdr1os loci was absent in the rat genome annotation. Thus, given diverged expression and sequence in circRNAs across species, it still requires a comprehensive understanding of circRNA species in rat, especially in tissues, development and diseases.
CNS-enriched circRNAs in RatNext, we focused on circRNA transcripts with a high abundance (BSJ read count ≥ 5) in a tissue identified from the rat body dataset and performed unsupervised clustering analysis. A total of 3231 circRNAs were clustered into six clusters and most circRNAs showed CNS- (46.2%), testis- (21.5%) and hemic/immune- (13.9%) enriched (Fig. 3A). Functional enrichment analysis of host genes generating those circRNAs in each cluster was performed. Host loci in cluster 1 were enriched in terms related with spermatogenesis (Fig. 3B). Analysis of tissue-specificity of circular and their cognate linear transcripts showed that most were testis-specific (tau index > 0.8) and the specificity of circular transcripts was greater than that in linear form (Fig. 3C). Six circRNA transcripts showed a higher expression than that in cognate linear transcripts, including circRNAs originating from Dock3 and Mtm1. Host loci (e.g., Satb1) generating circRNAs in cluster 6 were most enriched in terms related with immune, including myeloid cell differentiation (Fig. 3D and 3E). Host loci generating circRNAs in cluster 5 were enriched in tissue/organ development, including heart development, vasculature development and renal development (Fig. 3F). These loci included some tissue-specific genes, and most circRNAs originating from those loci were almost tissue-specific, such as Alb in liver and Ttn in heart (Fig. 3G). Ttn (Titin), encoding a giant protein, consisting of 349 exons in rat, generated 33 expressed circRNAs with 29 heart-specific and 2 skeletal muscle-specific (Fig. 3G and Supplementary file 1: Fig. S7). Of Ttn-derived circRNAs, the circRNA (Ttn-circ(143,144)) showed a similar abundance in between heart and skeletal muscle with a total BSJ read count of 105, respectively (Supplementary file 1: Fig. S7).
Fig. 3
CNS-enriched circRNAs in adult rat tissues. A The expression patterns of high abundant circRNAs in diverse rat tissues. B The top functional enrichment of host loci generating circRNAs in cluster 1. C Tissue-specificity of linear and circular forms of cluster 1 host loci. Red text indicated CIRCscore greater than 1. D The top functional enrichment of host loci generating circRNAs in cluster 6. E An example of immune-enriched circRNAs derived from Satb1. F The top functional enrichment of host loci generating circRNAs in cluster 5. G Examples of tissue-specific loci. Number in the bracket indicted the number of tissue-specific circRNAs. H The top functional enrichment of host loci generating circRNAs in CNS-enriched clusters 2, 3, and 4. I The loci generate multiple circRNAs and cluster information. Line width represents the number of circRNAs. J The expression percent of linear RNAs and circRNAs. Colorful lines and dots indicated that a higher CIRCscore in both rat and mouse orthologous circRNAs except a circRNA generating from a lncRNA (red star). K CircRNAs generated from Rere and expression in major rat tissues. L CircRNAs generated from Rims2 and expression in major rat tissues. M A conserved Rims2-dervied circRNA ortholog in mouse and its expression in different brain regions
Host loci generating circRNAs in CNS-enriched clusters were most enriched in terms of nervous system development, including synaptic signaling and neuron projection development (Fig. 3H). Analysis of loci that generated multiple circRNA transcripts (≥ 5) showed 14 synapse-related genes, including Rims1, Rims2 and Anks1b (Fig. 3I). Of 22 Rims2-derived circRNAs, most showed a higher expression in cerebellum than that in other CNS tissues. In general, most loci generate a major circRNA transcript. For example, Rims2-circ(20, 21, 22, 23) (namely e transcript in Fig. 3L) was the most abundance and showed a greater expression than the linear form. The orthologous exons formed BSJ of that circRNA in mouse (namely Rims2-circ(20, 21, 22)) also showed the highest expression in cerebellum and a higher expression than that in linear form (Fig. 3M). We also observed that some loci generated multiple major circRNA transcripts, such as Anks1b and Rere (Fig. 3K and Supplementary file 1: Fig. S2). Unlike Rims2-dervied circRNAs, most Rere-dervied circRNAs showed a higher expression in cortex and multiple Rere-dervied circRNAs also showed a higher expression than that in linear form especially in medulla (Fig. 3K). We next focused on those CNS-enriched circRNAs with a higher CIRCscore (> 1) and classified into three categories according to the percent of expression in CNS tissues compared to that in the total tissues (Fig. 3J). As observed, a higher percent of CNS expression of circular form than that in the linear form, such as a circRNA produced by a long non-coding RNA (ENSRNOG00000066491, here designed as lncRNA-66491, Fig. 3J). We also checked CIRCscore of mouse orthologs using a mouse body dataset[9] and some of them showed a higher expression and a higher CIRCscore in brain than that in other tissues, such as a circRNA generated from Boc (Fig. 3J). Taken together, we showed the CNS-enriched circRNAs as well as circRNAs enriched in other tissues in rat.
Developmental- and injured-regulated circRNAs in rat spinal cordAfter discussing circRNA expression in diverse tissues, we explored the changes in expression during central nervous system development and disease in rats. Due to a lack of long-term time-series RNA-Seq dataset for other rat CNS tissues, we used our developing spinal cord as well as injured spinal cord to investigate expression changes. First, we employed a regression-based method to detect significant expression profile difference of highly abundant circRNAs (BSJ read count ≥ 5 in at least two replicates) during the developing and injured spinal cord using the maSigPro package [21] respectively. We identified 1505 (out of 2289; 65.7%) developmental-regulated circRNAs generating from 979 loci and clustered into nine clusters (Fig. 4A and B). CircRNAs in most clusters presented a high expression in adult (P8w-P12w), except for the clusters 1 and 2. Functional enrichment of host loci generating circRNAs in clusters 1 and 2 were most enriched in the terms of “chromatin remodeling” and “axon guidance” respectively (Fig. 4B). A conserved circRNA (circ-Mettl9) from the cluster 2 with expression decrease along the development had been validated in mouse retinal development [13]. Host loci generating circRNAs in other clusters were most enriched in synapse or vesicle-mediated transport to the plasma membrane (Fig. 4B). Of other clusters, we noticed that circRNAs in the cluster 6 featured an increase at the neonatal stages (E18d ~ P3d) and parent host loci enriched in terms of “synapse organization” and “neuron projection development”, including Anks1b, Kcnk10, Nrcam, Mapk4, and Minar1 (Fig. 4B). Most abundant circRNAs were presented in the clusters 4 (e.g., circ-Gigyf2), 5 (e.g., circ-Crebrf) and 8 (e.g., circ-Ssbp2). Several studies have described a low correlation between circRNA and cognate linear RNA forms [29]. It was also consistent that a low correlation (rho: 0.13) between circRNAs and cognate linear RNA forms using the expression abundance from this dataset of developing rat spinal cord (Supplementary file 1: Fig. S8). We next asked which circRNAs showed a large difference between circRNAs and cognate linear RNA forms. We then calculated Euclidean distance between circRNAs and linear RNA forms and also considered the abundance of circRNAs and presented in Fig. 4C. We found several top abundant circRNAs that displayed a large divergence, including circ-Gigyf2, circ-Zranb1, circ-Ssbp2, circ-Fat3, and circ-Stau2 (Fig. 4C). Whether this characteristic also present using the dataset of injured spinal cord? We detected 240 injured-regulated circRNAs in long-term time-series of injured spinal cord (0.5 h (h) to 28 days post-injury, only rostral samples were considered). Most circRNAs were down-regulated, including circ-Gigyf2, circ-Fat3, and circ-Stau2 (Fig. 4D and Supplementary file 1: Fig. S9). The expression patterns and validation of circRNAs (e.g., circ-Akap6) have been described and validated in original publication of injured spinal cord [33]. Consistent observation in developing spinal cord, we also found some of those top abundant circRNAs (e.g., circ-Gigyf2, circ-Fat3, circ-Stau2 in Fig. 4E) showed divergent expression between circRNAs and cognate linear RNA forms after injury. For example, linear Gigfy2 showed a little change in expression pattern during the developing and injured rat spinal cord, while circ-Gigyf2 showed a dramatic increase in adult spinal cord and down-regulated after SCI (Fig. 4E). Taken together, we displayed the circRNAs expression patterns in rat spinal cord during the development and injury.
Fig. 4
Expression patterns of circRNAs in developing and injured spinal cord and divergence between circRNAs and linear RNAs. A The expression of developmentally regulated circRNAs. B Expression patterns of circRNAs and enrichment terms of related host loci. C The expression divergence between circRNAs and linear RNAs. Point colors depict clusters in A and B panels. Point sizes depict upper quartile of expression abundance. D Expression patterns of 20 abundant circRNAs in spinal cord after injury (identified injury-related circRNAs were shown in Supplementary file 1: Fig. S9). “Avg” panel depict average of expression abundance. Pink star indicated validated expression in spinal cord after injury. Red star indicated documented exploration of function. E Three examples of circRNAs with expression diverged from that in linear RNAs during the developing and injured spinal cord
Experimental validation of circular RNAs in developing spinal cord and other tissuesWe screened 12 circRNAs for experimental verification based on their abundance and expression changes across various tissues and during spinal cord development, including circRNAs derived from Ssbp2 and Minar1. To verify the circular structure of these circRNAs, total RNA isolated from rat spinal cord was treated with RNase R, an exonuclease that selectively degrades linear RNAs. Following RNase R treatment, PCR amplification using divergent primers, followed by agarose gel electrophoresis, yielded a detectable product specific to the circRNA (Fig. 5A). In contrast, the linear RNA (Gapdh) was vanished in the RNase R-treated sample, supporting the circular nature of the circRNAs. Subsequent Sanger sequencing of the PCR products confirmed the presence of the back-splicing junction (Fig. 5B), providing further evidence for the circular configuration of circRNAs. The above results validate the 12 circRNAs screened from the RNA sequencing datasets by RNase R treatment and Subsequent Sanger sequencing.
Fig. 5
Experimental validation of 12 circRNAs and relative expression in diverse rat tissues and developing spinal cord. A Agarose gel electrophoresis assay confirmed the resistance to exonucleases of 12 candidate circRNAs by RNase R treatment. Linear Gapdh was used as the negative control. The uncropped electrophoresis image was shown in Supplementary file 1: Fig. S11. B Sanger sequencing result of qPCR products confirmed the back-splicing junction sequences from 12 candidate circRNAs. Arrows in the upper of base sequences indicated the back-splicing site. C Relative expression of 12 candidate circRNAs in six tissues of rats. Data are present as mean + S.E.M. (n = 3 samples per group, *p < 0.05, unpaired t-test; #p < 0.05, one-way ANOVA test). D Relative expression of 12 candidate circRNAs in five developmental timepoints of rat spinal cord. Data are present as mean + S.E.M. (n = 3 samples per group)
To assess the relative expression of these circRNAs, we conducted a qPCR experiment across six tissues (cortex, hippocampus, spinal cord, heart, liver, kidney) and five developmental timepoints (E11d, E18d, P1d, P2w, P8w) (Fig. 5C and 5D). All of these circRNAs exhibited significantly higher expression levels in CNS tissues (cortex, hippocampus, and spinal cord) compared to non-CNS tissues (heart, liver, kidney), such as circ-Anks1b, circ-Nrcam, circ-Minar1, and circ-Ssbp2 (Fig. 5C, p < 0.05, unpaired t-test). In addition, four circRNAs showed significantly differential expression across CNS tissues (Fig. 5C, p < 0.05, one-way ANOVA test). For example, circ-Minar1, consisting of a single exon, showed the highest expression in the rat spinal cord and was also highly expressed in the mouse spinal cord (Supplementary file 1: Fig. S10). Generally, these circRNAs had low expression at the early embryonic stage (E11d) and increased during later embryonic and neonatal stages (E18d and P1d), except for circ-Brsk2, which expressed only after adolescence (P2w) and increased with development. In summary, our experimental validation confirmed the circRNAs predicted from RNA sequencing datasets and their generally consistent expression patterns across tissues and developmental stages.
Functional exploration of abundant circRNAs in rat modelAfter confirming the accuracy of circRNA predictions, we explored their potential functions by integrating other omics datasets and bioinformatic predictions. The circRNAs function in various biological processes, including acting as sponges for miRNAs or RNA-binding proteins (RBPs) and translating into peptides or proteins [8]. We used ribosome footprint profiling (RFP), polysome profiling, and mass spectrometry to investigate the translation potential of circRNAs. Due to short fragment lengths in RFP (28–35 nt), translated circRNAs are often underestimated [11, 37]. We re-analyzed ribosome footprint and polysome profiling data from rat hippocampus [38]. As expected, no ribosome-associated circRNAs were detected in the RFP datasets using the same pipeline. However, we identified 8350 credible circRNAs (BSJ read count ≥ 5 in both two replicates), with 8280 (99.16%) in ribosome-free, 1186 (14.28%) in monosome-associated, and 123 (1.47%) in polysome-associated fractions (Fig. 6A). The ratio of expression between monosome- and polysome-associated circRNAs was higher for circRNAs and lower for linear RNAs (Fig. 6A), indicating that most circRNAs are not translated, and those that are tend to be associated more with monosomes [39]. We investigated the top 20 abundant polysome-associated circRNAs, including circ-Ssbp2, circ-Cdyl, circ-Nfix, and circ-Hipk3 (Fig. 6C and Fig. 6D). Most of these circRNAs were also highly abundant in the developing spinal cord, except for circRNAs from Grm4 and Boc (Fig. 6C). Using IRESfinder and SRAMP, we predicted internal ribosomal entry sites (IRES) and N-6-methyladenosine (m6A) sites, finding that most of these circRNAs possessed open reading frames (ORF), IRES, and/or m6A sites (Fig. 6C), suggesting their potential for translation.
Fig. 6
Functional exploration of candidate circRNAs by integrating multi-omics datasets and in silico. A The summary of the number of circRNAs identified from polysome profiling dataset of rat hippocampus. B The ratio between monosome- and polysome- expression in circRNAs and cognate linear RNAs. C The expression of top 20 polysome-associated circRNAs. D The BSJ coverage of four circRNAs as well as predicted the longest translated protein. E Three miRNAs may bind to the pseudo circ-Coq3. F High abundant single-exon derived circRNAs with multiple miRNAs binding. G The conserved circ-Fat3(2) and predicted miRNA binding sites. Colorful rectangles showed the top 10 abundant miRNAs, and grey rectangles showed other miRNA binding sites. The top 10 abundant miRNAs were labelled
Next, we investigated the role of highly abundant circRNAs in the developing spinal cord as miRNA sponges. We considered only miRNAs with a maximum number of reads greater than 50 in at least two replicates (Fig. 1A). We scanned for miRNA binding sites spanning or near the back-splicing junction of circRNAs (pseudo-circRNAs) or within single-exon derived circRNAs. This analysis included 364 miRNAs and 2289 circRNAs (176 single-exon derived). We predicted 416 (194 miRNAs and 316 circRNAs) and 1522 (256 miRNAs and 167 circRNAs) miRNA-circRNA interactions for pseudo-circRNAs and single-exon derived circRNAs, respectively, with support from three tools (Supplementary file 4: Table S3).
We observed that miR-351-5p and miR-125a/b-5p may bind to circRNAs generated from Coq3 (Coenzyme Q3, Fig. 6E). Among single-exon derived circRNAs, eight with a total BSJ read count greater than 1,000 had multiple miRNA binding sites, including circ-Fat3(2), circ-Slc8a1(2), and circ-Cdyl(2) (Fig. 6F). Circ-Fat3(2), generated from exon 2 of rat Fat3 loci (orthologous to “mmu_circ_0001746” in mouse), is evolutionarily conserved and has been implicated in neural development in mice[40]. We predicted 25 miRNAs targeting circ-Fat3(2), supported by three miRNA target prediction tools, including miR-125b-2-3p, miR-146b-5p, miR-133a-3p, and miR-1224 (Fig. 6F and Fig. 6G). These analyses suggest that circRNAs may act as miRNA sponges and potential translational templates, highlighting their diverse roles in gene regulation.
Overview of analyzed datasets and accessible resourceOverall, we re-analyzed a total of 22 RNA-Seq datasets from 20 publications[9,10,11,12,13, 15, 18,
Comments (0)