Antibodies and reagents
The primary antibodies employed in this investigation consisted of: anti-PRDM16 (Cat# PA5-20872, Thermo Scientific), anti-β-actin (Cat# 66009-1-Ig, Proteintech), anti-USP10 (Cat# 19374-1-AP, Proteintech), anti-GSDME (Cat# ab215191, Abcam), anti-HA-Tag (Cat# 3724, CST), anti-Caspase-3 (Cat# 9662, CST), anti-Cleaved Caspase-3 (Cat# 9664, CST), anti-NLRP3 (Cat# T55651, Abmart), anti-GSDMD (Cat# 20770-1-AP, Proteintech), anti-Cleaved GSDMD (Cat# 10137, CST), anti-Cleaved Caspase-1 (Cat# 89332, CST), anti-CD62p (Cat# bs-0561R, Bioss). Secondary antibodies were procured from Affinity (Wuhan, China). An enhanced chemiluminescence assay kit (Cat# 180–5001, Tanon, Shanghai, China) was used for protein band detection. USP10 plasmid, PRDM16 shRNA, and USP10 shRNA were procured from Genechem (Shanghai, China). Doxycycline (DOX) was sourced from Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. PLGA 50:50 (Cat# MB5649-1, Meilunbio) and formononetin were obtained from the Guangzhou Institute of Biomedicine and Health, Guangzhou, China.
Cell culture and treatments
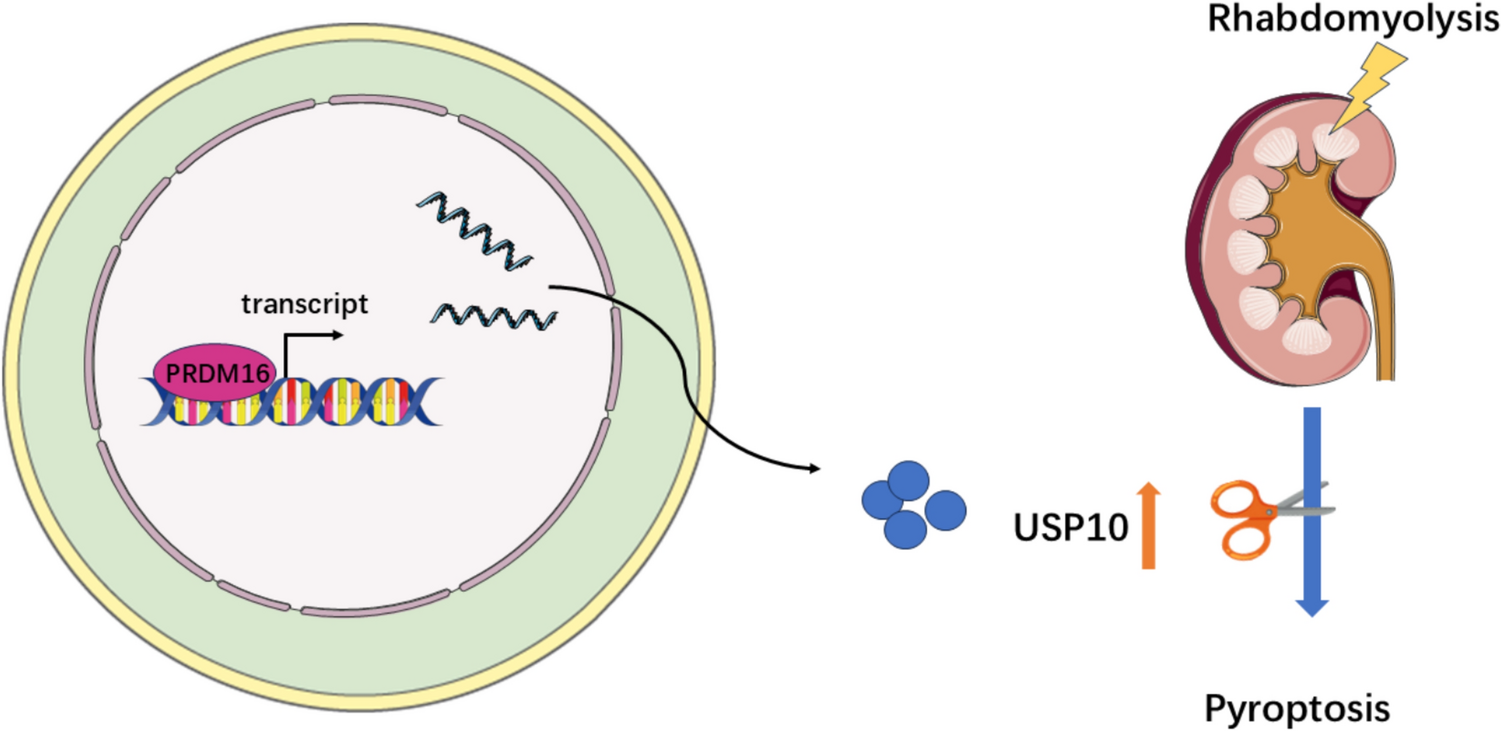
The BUMPT cell line (Boston University mouse proximal tubular cells) was kept in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 0.5% penicillin, and streptomycin, and incubated in a 5% CO2 atmosphere at 37 °C [17]. A DOX-induced HA-PRDM16 stable cell line was established following our previous protocol [18], using 500 ng/ml of doxycycline. Cells underwent transfection with PRDM16 shRNA (100 nM), USP10 shRNA (100 nM), USP10 plasmid (1 μg/ml), or scramble shRNA and empty plasmid utilizing Lipofectamine™ 2000 (Thermo Scientific, USA). The cellular model of RM-AKI was established as described previously [10, 19], where BUMPT cells were incubated with 200 mM ferrous myoglobin for 24 h. Cells were pretreated with Disulfiram (0.5 μM, HY-B0240, MCE, China) and Z-DEVD-FMK (100 μM, HY-12466, MCE, China) for 2 h prior to experimentation.
Animal model of rhabdomyolysis-induced AKI
All animal experiments were executed per the protocols set by the Animal Care Ethics Committee of the Second Xiangya Hospital of Central South University (No. 2018065). Steps were taken to reduce animal usage and minimize their distress. Eight-to-ten-week-old male C57BL/6J mice were procured from SJA Laboratory Animal Ltd (Changsha, China) and kept in the Animal Experimental Center of the Second Xiangya Hospital. Mice were anesthetized using sodium pentobarbital. The proximal tubule-specific PRDM16-knockout (PT-PRDM16-KO) and PRDM16 knock-in (PT-PRDM16-KI) mice were generated as previously described [18]. These mice were kept under regulated conditions with alternating 12-h light/dark periods and unrestricted food and water availability. To induce RM-induced acute kidney injury (RM-AKI), mice received a 7.5 mL/kg injection of 50% glycerol diluted in sterile water into the skeletal muscles of the hind limbs bilaterally, as previously reported [20]. The control group received an equivalent amount of normal saline. Mice were sacrificed at 12- and 24-h post-injection, and kidney tissue and blood samples were collected for analysis. PNPs encapsulated with 20 mg/kg formononetin were administered via tail vein injection 30 min following RM-AKI induction.
Assessment of cell viability and LDH
Cells were placed at 5000 cells/100 μL in 96-well plates. After treatment, cell viability and LDH levels were assessed utilizing the Viability/Cytotoxicity Multiplex Assay Kit (CK17, Dojindo, Japan) per the supplier’s protocol. Absorbance was ascertained utilizing a Spark® Multi-function Microplate Detector (TECAN, Switzerland).
Flow cytometry detection of ROS
Cellular reactive oxygen species (ROS) levels were ascertained utilizing the Reactive Oxygen Species Assay Kit (MA0219, Meilunbio, China). The fluorescence intensity change of the DCFH-DA (2,7-Dichlorod-hydrofluorescein diacetate) probe was utilized to quantify intracellular ROS levels. Fluorescence intensity was measured by flow cytometry (Cytek, USA) according to the kit protocol.
Relative quantitative real-time PCR (RT‒qPCR)
Total RNA was procured from the renal cortex or BUMPT cells utilizing RNAiso Plus reagent (Cat# 9108, Takara, Japan). cDNA was generated utilizing the Evo M-MLV Reverse Transcription Kit (Cat# AG11705, Accurate Biology, China). RT-PCR quantification was executed utilizing SYBR Green Pro Taq HS reagent (Cat# AG11702, Accurate Biology, China) and a LightCycler 96 system (Roche). Relative quantification was calculated utilizing the ΔΔCT technique. The primer sequences employed for RT-qPCR were as follows: (Mouse) β-actin: forward Primer 5′-GGCTGTATTCCCCTCCATCG-3′, reverse Primer 5′-CCAGTTGGTAACAATGCCATGT-3′; (Mouse) PRDM16: forward Primer 5′-CAGCACCTCCAGCGTCACATC-3′, reverse Primer 5′-GCGAAGGTCTTGCCACAGTCAG-3′; (Mouse) USP10: forward Primer 5′- AACCCACAGTATATCTTTGGCG-3′, reverse Primer 5′-CCCTCACTAGGTTCGATGACTTC-3′.
Western blotting (WB)
Protein extracts from mouse renal cortex or BUMPT cells were procured utilizing RIPA buffer comprising protease inhibitors. Equal amounts of protein samples underwent SDS-PAGE fractionation and were subsequently blotted onto Polyvinylidene difluoride (PVDF) membranes. The membranes underwent blocking with 5% skim milk and subsequent overnight exposure to primary antibodies at 4 °C. Following PBST washes, the membranes were exposed to horseradish peroxidase (HRP)-linked secondary antibodies. Protein band detection was accomplished utilizing NcmECL Ultra (Cat# P10200, NCM Biotech, China). WB images were captured using a gel documentation system (Tanon, China), and band density quantification was performed using ImageJ (version 1.54 g).
Immunofluorescence staining
Immunofluorescence staining of BUMPT cells was performed as per the manufacturer's instructions (Cat# P0183, Beyotime, China). Cells grown on coverslips were incubated with PRDM16 antibody (1:500 dilution) overnight at 4 °C, succeeded by exposure to the secondary antibody for 1 h at ambient temperature. Cell nuclei underwent DAPI staining for 3–5 min. Immunofluorescence images were procured utilizing an Eclipse Ti laser scanning confocal microscope (Nikon, Japan), and relative fluorescence intensity was quantified using ImageJ software (version 1.54 g).
ChIP analysis
Chromatin immunoprecipitation sequencing (ChIP-seq) data utilized in this investigation were obtained from two sources. The first part of the data was from our previous study, which involved ChIP-seq analysis of a DOX-induced HA-PRDM16 stable cell line [18]. The second part was procured from the Gene Expression Omnibus (GEO) database, with the access number GSE131487. This dataset includes ChIP-seq data from the intestine tissue of control and PRDM16-knockout mice [21]. All ChIP-seq data were depicted through Integrative Genomics Viewer (IGV, version 2.18.4) [22]. The binding sites and motifs of PRDM16 in the USP10 promoter region were predicted using the online tool MoloTool [23]. ChIP PCR and ChIP qPCR experiments were performed per the protocol outlined in the ChIP Assay Kit (Cat# P2078, Beyotime, China). The primers used in these experiments were designed using NCBI's online tool (primer blast), and the target amplified fragments contain the predicted binding sites. The primers are as follows: P1 Forward primer 5′-TCCCCAATGCCAGGTAAAACA-3′, Reverse primer 5′-TCCCCAATGCCAGGTAAAACA-3′; P2 Forward primer 5′-GTACTAGGGAAGTAGGGGCGG-3′, Reverse primer 5′-TGTAAAGCCCCAGCTGTCCTG-3′.
Measurement of IL-18, IL-1β, BUN, and creatinine
ELISA experiments for the detection of cytokines were performed using the Mouse IL-1β ELISA Kit (Cat# PI301, Beyotime, China) and Mouse IL-18 ELISA Kit (Cat# PI553, Beyotime, China). BUN and creatinine levels were measured using the Urea-Nitrogen-Content Assay Kit (Beijing Boxbio Science & Technology) and Creatinine (Cr) Assay Kit (sarcosine oxidase) (Nanjing-Jiancheng Bioengineering Institute, Nanjing, China), respectively.
HE, TUNEL, DHE staining, immunohistochemical staining, and qualification
Histological damage was assessed using Hematoxylin and eosin (H&E) staining. Tubular damage [24] was scored utilizing the percentage of injured tubules, with the following grading scale: 0% (0); < 25% (1); 25%–50% (2); 50%–75% (3); and > 75% (4). Four fields chosen at random were evaluated, and their mean score was determined. TUNEL staining was executed utilizing the TUNEL kit (Cat # G1502, Servicebio, China), and the number of TUNEL-positive cells was counted in 10–20 randomly selected fields per tissue section. Dihydroethidium (DHE) staining was employed to examine ROS levels in kidney tissue. Renal cryosections were incubated with DHE solution (Cat #D7008, Sigma Aldrich, Germany) for 1 h, washed, and examined under fluorescence microscopy. The DHE fluorescence intensity was analyzed using ImageJ software (version 1.54 g). Four arbitrarily chosen fields were scored, and the geometric mean fluorescence intensity was calculated. Immunohistochemical (IHC) analysis was performed as described in previous studies [25]. Specimens of tissue underwent overnight incubation at 4 °C utilizing primary antibodies (PRDM16, diluted at 1:100). IHC scores were semi-quantitatively assessed by computing the product of staining strength and the percentage of cells exhibiting positive staining [26].
Preparation of PNPs
Platelet membrane-coated nanoparticles (PNPs) were prepared using emulsified solvent evaporation and ultrasonication techniques, as described in previous studies [27]. Initially, poly(lactic-co-glycolic acid) (PLGA)-encapsulated formononetin was prepared following the methods outlined in our previous research [16]. Platelet membranes were then isolated from mice according to a previously established protocol [28]. In the final step, PLGA-encapsulated formononetin and the pre-prepared platelet membranes were sonicated at 30% amplitude for 10 min to form the platelet membrane-coated nanoparticles (PNPs). The structural features of the microspheres were examined through Transmission Electron Microscopy (TEM, HITACHI HT7800, Japan). The size distribution of the microspheres was evaluated utilizing a Nanoparticle Tracking Analyzer (NTA, Particle Metrix, Germany), with measurements performed three times to verify reproducibility. The electrical surface properties (zeta potential, mV) of the nanoparticles were assessed by employing a Zetasizer Nano ZS particle analyzer (Malvern Instruments).
Statistical analysis
Statistical analysis was executed employing SPSS (version 25.0) and GraphPad Prism (version 9.0) software. Results are denoted as means ± standard deviation (SD). Statistical comparisons between two groups were examined via two-tailed Student’s t-tests, whereas multiple group comparisons utilized one-way ANOVA. Statistical significance was set at P-values < 0.05.



Comments (0)