
Remember me
A total of 27 patients with histologically confirmed breast or prostate cancer and osteoblastic or mixed bone lesions took part in this phase I/II study. All patients had symptomatic bone metastases verified by [99mTc]Tc-MDP scans, a Karnofsky performance status ≥ 50%, and an expected survival of at least 3 months. The characteristics of the study population are summarized in Table 1.
Table 1 Characteristics of study populationStudy design and groupsParticipants were split into two major groups randomly:
1.Pharmacokinetics and Biodistribution Group
This group included 17 patients and was further subdivided into two subgroups:
In this sub group, patients were divided into groups of three and injected with escalating activities of [177Lu]Lu-EDTMP. Specifically, the administered activity was designed to deliver estimated bone marrow doses equivalent to 75%, 100%, and 125% of the 200 cGy reference value for bone marrow, as predicted by the "test dose" study calculations described below.
Blood/urine sampling, plus gamma camera imaging at multiple intervals (30 min, 4 h, 8 h, 24 h, 48 h, 7 days, 14 days).
Bone uptake was calculated using Excel-based software for quantitative assessment.
Low activity of [177Lu]Lu-EDTMP (1.3 GBq, 35 mCi), or
High activity of [177Lu]Lu-EDTMP (2.6 GBq, 70 mCi).
2.Clinical Efficacy and Toxicity Group
This group comprised 18 patients, including 8 patients from Subgroup 2 of the pharmacokinetics and biodistribution study, who had already received either low or high activity, and 10 additional new patients. The patients were further divided into two arms:
Arm 1 (n = 9): Received low activity of [177Lu]Lu-EDTMP (1.3 GBq, 35 mCi).
Arm 2 (n = 9): Received high activity of [177Lu]Lu-EDTMP (2.6 GBq, 70 mCi).
Inclusion and exclusion criteriaInclusion CriteriaPatients meeting the following criteria were eligible for the study:
Histologically confirmed diagnosis of breast or prostate cancer.
Hormone refractory prostate cancer, defined as:
Two consecutive rises in serum Prostate-Specific Antigen (PSA) done measured 1 week apart, or
Disease progression on [99mTc]Tc-MDP bone scintigraphy, with the appearance of ≥ 2 new lesions.
Breast cancer with bone metastases.
Predominantly osteoblastic skeletal metastases as demonstrated by increased uptake (“hot foci”) on a [99mTc]Tc-MDP bone scan performed within 4 weeks prior to [177Lu]Lu-EDTMP administration. Painful sites described by the patient must correlate with the hot foci on the bone scan.
Life expectancy ≥ 3 months.
Karnofsky’s index ≥50%
Written informed consent, provided by the patient.
Exclusion criteriaPregnancy and breastfeeding.
Risk of pathological bone fracture requiring immediate intervention.
Previous External Beam Radiation Therapy on >30% of the bone marrow.
Hematological values outside the following limits:
Hemoglobin (Hb) < 10 g/dL.
White blood cell count (WBC) < 3 × 10⁹/L.
Neutrophils < 1.5 × 10⁹/L.
Platelets < 100 × 10⁹/L.
Serum bilirubin > 1.5 times the upper normal limit (UNL).
Serum creatinine > 2 times the UNL.
Previous radiotherapy or chemotherapy within the last 4 weeks.
Prior radiopharmaceutical therapy within the previous 2 years.
Extensive metastatic disease demonstrated as a superscan on bone scintigraphy.
Presence of Central Nervous System or epidural metastases.
Any life-threatening comorbid illness.
History of bleeding disorders that could be exacerbated by the study medication.
Concurrent illnesses that could interfere with study results or completion.
Patients undergoing neural block treatments for bone pain.
Patient preparation and injectionFor the escalating activity schedule, a tracer activity of 75 MBq (2 mCi) of [177Lu]Lu-EDTMP was administered, and whole-body scans were performed within 1 h (pre-void) and at 24 h post-injection. The desired red marrow absorbed dose was determined based on the bone marrow estimated dose of 100 cGy, 150 cGy, 200 cGy, 250 cGy, and 300 cGy.
Patient-specific parameters, including body height, weight, and desired red marrow absorbed activity, were incorporated into an Excel-based software program to calculate the therapeutic activity for each patient. The calculated activity, measured using an activity calibrator, was administered via a three-way cannula after ensuring adequate hydration and recording baseline vital signs. The injection procedure involved slow administration of the radiopharmaceutical over 30 s, followed by a 10 mL saline flush. Pre- and post-injection syringe activities were measured to determine the actual injected activity. Vital signs were monitored every 30 min for 2 h post-injection to ensure patient safety.
ImagingWhole-body images were acquired using a dual-head gamma camera (GE Infinia with XelerisTM workstation, GE Healthcare, Milwaukee, WI, USA) in both anterior and posterior projections at 30 min (pre-void), 4 h, 8 h, 24 h, 48 h, 7 days, and 14 days after [177Lu]Lu-EDTMP injection. The scans at 7 and 14 days were conducted primarily for visual analysis to observe the retention of activity. For analytical purposes, only data from the 4 h, 24 h, and 48 h time points were used.
The imaging parameters included the use of a medium energy general-purpose (MEGP) collimator, a scan speed of 10 cm/min, a total scan length of 200 cm, and a total scan time of approximately 20 min. The energy window was centered at 208 keV with a ± 7.5% margin. The matrix size used was 256 × 1024 with a zoom factor of 1, and body contouring was disabled. To maintain consistency, the position of head 1 was noted for each patient to standardize imaging conditions across time points. All images were digitally stored for post-processing and further analysis.
Blood and urinary clearanceEstimation of blood clearanceBlood clearance was assessed using serial blood samples of 2 mL each, collected at 2 min, 4 min, 8 min, 15 min, 30 min, 2 h, 4 h, 8 h, and 24 h post-injection. Blood samples were prepared in 10 tubes, each filled with 100 µL of blood using a heparinized syringe. The tubes were weighed before and after filling using an electronic balance to determine the exact volume. Radioactivity in the blood samples was measured using a gamma counter (Packard Cobra II Auto Gamma Counter, Packard Instrument Company—now part of PerkinElmer, US), and the total blood activity was normalized as a percentage of the injected activity to generate the blood clearance curve.
Determination of urinary clearanceUrine was collected over a 48-h period and fractionated into the following intervals: 0–4 h, 4–8 h, 8–24 h, and 24–48 h. For each interval, two vials of 10 mL urine were prepared and weighed before and after filling to determine the volume accurately. Activity in each sample was measured using a dose calibrator (Capintec CRC-15R Dose Calibrator, Capintec, Inc., US). Urinary excretion was calculated as a percentage of the total administered activity using the following formula:
Ap: percentage of injected activity in urine at time (t).
At: Activity in urine at time (t) × 100.
TT: Total administered activity.
Determination of bone uptakeWhole body count method (Excel-Based Software)Patient-specific data, including age, gender, height, weight, and desired red marrow absorbed dose, along with background-corrected whole-body counts (anterior and posterior projections at 4 h, 24 h, and 48 h), were entered into an Excel-based program. Bone uptake at each time point was expressed as a percentage of the 30-min counts, defined as 100%. The mean values of bone uptake were calculated for further analysis.
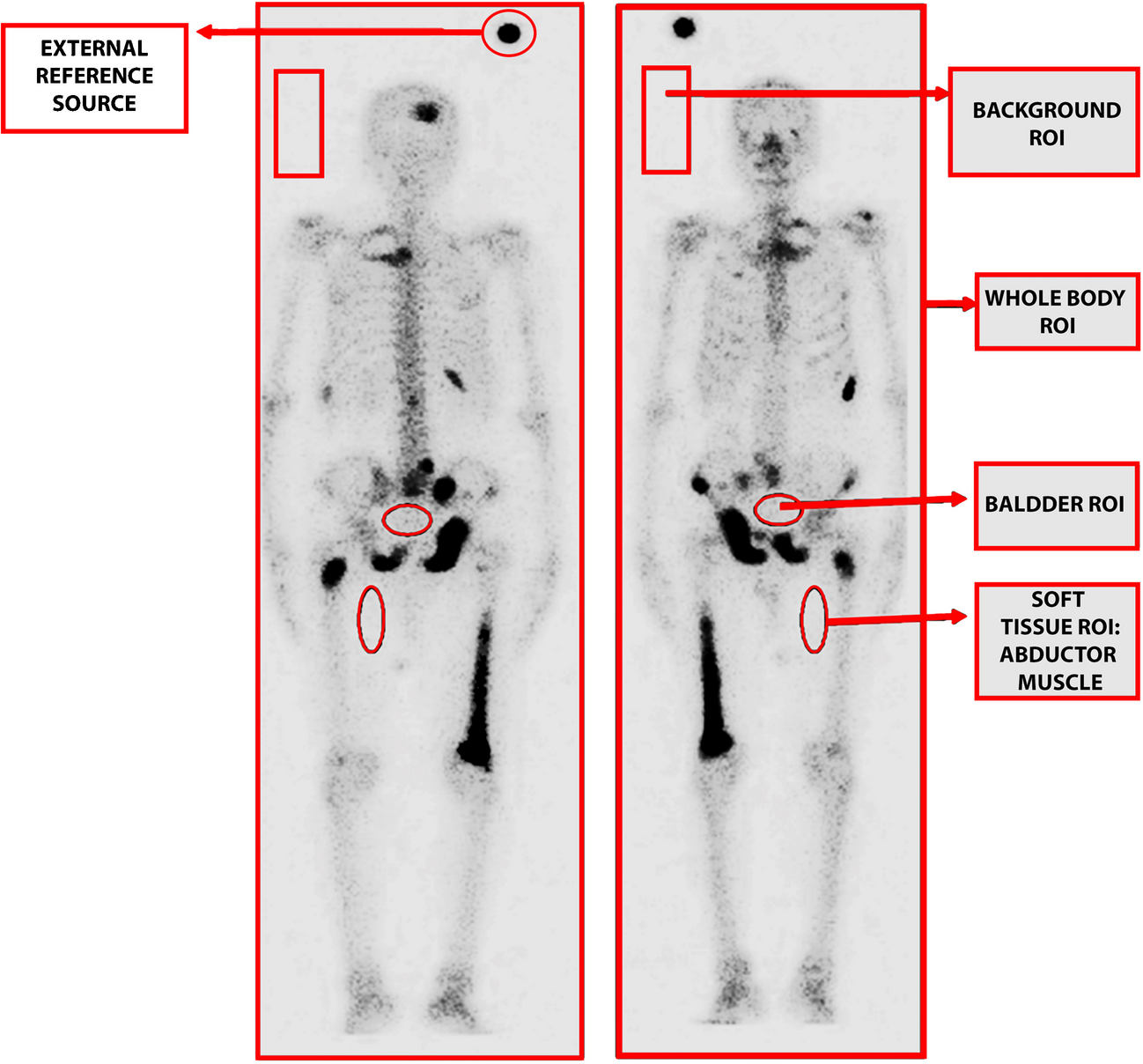
Scintigraphic methodFor each image, the activity levels of the whole body, urinary bladder, and adductor muscle of the thigh (representing soft tissue) were measured using a conventional region-of-interest (ROI) technique applied to both anterior and posterior projections. Additionally, a rectangular ROI was placed adjacent to the head and shoulders to account for bremsstrahlung radiation from the β-particles of the radionuclide. An example of ROI positions is provided in Fig. 1.
Fig. 1
Regions of Interest (ROIs) for determination of bone uptake, soft tissue retention, and urinary excretion of [177Lu]Lu-EDTMP by Gamma Camera imaging
Using these measurements, the geometric mean for each ROI was calculated after applying corrections for radioactive decay, bremsstrahlung radiation, and pixel normalization. The total whole-body activity measured at 30 min post-injection was set as the baseline (100%) for each patient and served as the reference for subsequent activity calculations, expressed as percentages of this baseline value. At the 30-min time point, the whole-body activity was also considered the maximum soft tissue activity. In subsequent images, the activity of the adductor muscle was used as a marker of soft tissue retention over time. The ratio of soft tissue activity to the initial whole-body activity was therefore equivalent to the ratio of adductor muscle activity to its initial value at 30 min post-injection.
Urinary excretion was calculated by subtracting the whole-body activity at each subsequent time point from the baseline 30-min activity and adding the urinary bladder activity. Bone uptake at each time point was determined by subtracting both urinary excretion and soft tissue retention from the initial whole-body activity (100%).
Formulae for calculations are given below:
St: Soft tissue retention at time (t).
WT: Whole body counts at 30 min.
At: Adductor’s counts at time (t).
AT: Adductor’s counts at 30 min
Ut: Urinary excretion at time (t).
WT: Whole body counts at 30 min.
WB: Whole body counts at time (t).
Bt: Bladder’s counts at time (t)
Bt: Bone uptake at time (t).
WT: Whole body counts at 30 min.
St: Soft tissue retention at time (t).
Ut: Urinary excretion at time (t).
Uptake ratios between metastatic lesion, normal bone, and soft tissueEqual-sized ROIs were drawn over metastatic lesions, normal bone, and the adductor muscle of the thigh to calculate uptake ratios, as shown in Fig. 2. The ratios included lesion-to-normal bone, lesion-to-soft tissue, and normal bone-to-soft tissue. Data were analyzed at 30 min, 4 h, 8 h, 24 h, and 48 h.
Fig. 2
Regions of Interest (ROIs) for Determination of uptake ratios between metastatic lesions, normal bone, and soft tissue
Correlation studiesThe correlation between injected activity (MBq) and percent bone uptake was analyzed by plotting scatter graphs with injected activity on the x-axis and bone uptake percentages (at 4, 24, and 48 h) on the y-axis. Spearman correlation coefficients and p-values were calculated to determine statistical significance. Similarly, the bone lesion score, derived from [99mTc]Tc-MDP scans, was correlated with the percent bone uptake at the same time points.
Safety and toxicity profilePatients were monitored over a 12-week period to assess safety and toxicity. Hematological parameters, including Hb levels, white blood cell counts, and platelet counts, were closely monitored along with renal and liver function tests. Assessments were conducted at 1, 2, 4, 8, and 12 weeks. For prostate cancer patients, serum PSA levels were repeated at week 8.
Pain response assessmentPain response was evaluated through multiple parameters, including the Visual Analog Scale (VAS), type and frequency of analgesics, mobility, and the Karnofsky Performance Status (KPS).
Statistical analysisMean values and standard deviations were calculated for all measured parameters, including blood clearance, urinary excretion, and bone uptake. Correlation coefficients (Spearman's r) and p-values were computed to assess the relationships between variables. Pain response was analyzed using analysis of variance (ANOVA) to determine significant reductions in VAS scores and analgesic usage over time.
Data availabilityThe datasets generated during and/or analyzed during the current study are available from Dr Shabana Saeed, responsible of data collection and analysis, on reasonable request.
Human ethics and consent to participateThe research was undertaken in compliance with the Helsinki Declaration. Formal written patient consent was not required for this type of study. The study was approved by the Pakistan Institute of Engineering and Applied Sciences (P.I.E.A.S.), the Ethical Review Committee of the Nuclear Medicine, Oncology, and Radiotherapy Institute (N.O.R.I.)
Comments (0)