
Remember me
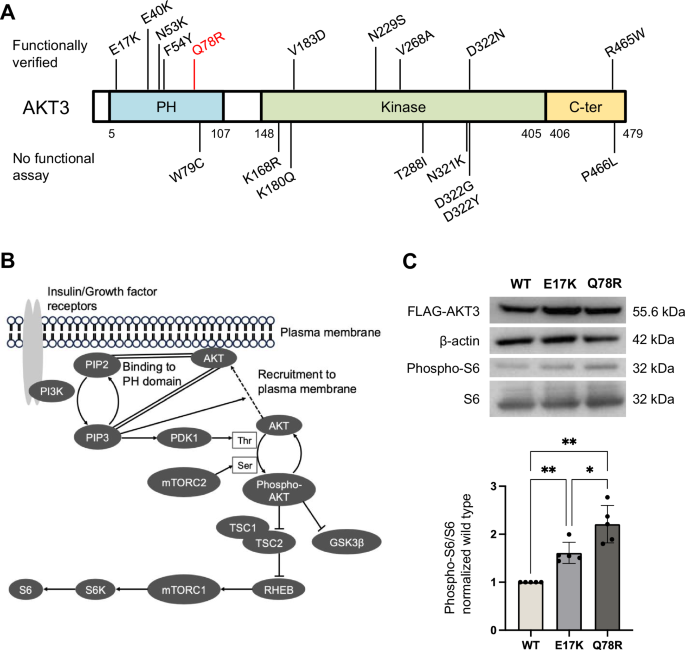

This study included 59 patients from 59 families (36 boys and 23 girls) suspected with CHARGE syndrome, with an average age at evaluation of 3 years and 2 months (range 0 months to 19 years). Patients were classified according to the Verloes and Hale criteria [5, 6], as illustrated in Fig. 1. Of the 59 patients, 44 (74.6%) were diagnosed with CHARGE syndrome according to the Verloes criteria, and 40 patients (67.8%) met the Hale criteria. A total of 38 patients met both criteria, whereas 13 patients suspected with CHARGE syndrome did not meet either the Verloes or Hale criteria.
Fig. 1
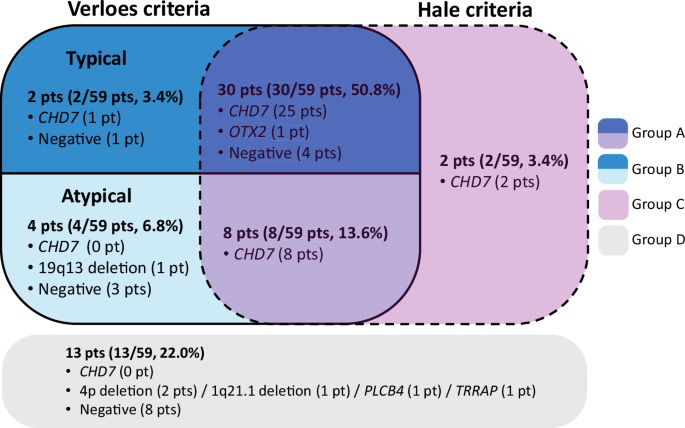
Classification of subjects based on Verloes and Hale criteria. Subjects were categorized based on their fulfillment of the Verloes (square box with bold border) and Hale criteria (square box with dashed border); they were divided into typical, atypical. CHARGE syndrome with different colors based on the Verloes criteria. Those meeting both criteria are represented in the overlapping section (Group A), those meeting only the Verloes criteria are in Group B, those meeting only the Hale criteria are in Group C, and those meeting neither criterion are shown in the gray zone (Group D) outside both criteria. Each section shows the number of subjects and frequency expressed as percentages. Cases where a causative variant was identified were noted as “gene name (number of subjects),” and cases without identified variants were marked as “negative (number of subjects)”
Patients were categorized as follows: those meeting both diagnostic criteria [Group A, 38 patients (64.4%)], those meeting only the Verloes criteria [Group B, 6 patients (10.2%)], those meeting only the Hale criteria [Group C, 2 patients (3.4%)], and those meeting neither criteria [Group D, 13 patients (22.0%)]. The phenotypes of the patients are detailed in Table 1 and Supplementary Table 1. The frequency of phenotypes for each group was analyzed, i.e., those meeting at least one diagnostic criterion (Groups A–C) and the entire cohort (Groups A–D), and the obtained frequency was consistent with that reported in previous studies [3, 6, 21,22,23,24,25,26,27] (Table 1).
Table 1 Frequency of clinical manifestations in patients suspected with CHARGE syndrome: A comparative analysis with previous literatureMajor criteria including coloboma, choanal atresia, and ear anomalies were highly frequent (> 70%), with ear anomalies present in up to 81.4% of the entire cohort (Groups A–D) and in 91.3% of those fulfilling either the Verloes or Hale criteria (Groups A–C). In the minor criteria, hearing impairment, short stature, cardiac malformation, and structural brain anomalies were also highly frequent ( > 70%). However, choanal atresia in the major criteria, anosmia, growth hormone deficiency, tracheoesophageal fistula, cleft lip or palate, renal anomalies, and skeletal or limb anomalies in the minor criteria were observed at a lower frequency ( < 30%), differing from previous reports.
Detection of CHD7 pathogenic variantsThe results of genetic testing are shown in Fig. 2. Among the 59 patients, 36 (60%) harbored pathogenic variants in CHD7 as evaluated by single-gene sequencing or MLPA analysis (listed in Supplementary Table 1 and Supplementary Table 2). The detection of CHD7 pathogenic variants varied by group classification as follows: Group A (33/38, 86.8%), Group B (1/6, 1.7%), Group C (2/2, 100%), and Group D (0/13, 0%). Of those who met either the Verloes or Hale criteria (Groups A–C), 78.3% (36/46) harbored pathogenic variants in CHD7.
Fig. 2
Analysis of pathogenic variants in subjects with clinically suspected CHARGE syndrome. n, number; MLPA, multiplex ligation-dependent probe amplification; CMA, chromosome microarray; ES, exome sequencing; GS, genome sequencing. A In the 59 patients suspected with CHARGE syndrome, single-gene sequencing or MLPA of CHD7 revealed pathogenic variants in 36 patients, representing 61.0% (36/59). This frequency is displayed in a pie chart differentiating between detected and not detected cases. B For the 23 patients where no variants were found in CHD7, additional genetic testing was conducted. A bar graph illustrates each additional testing method on the x-axis, with the number of tests performed indicated on the y-axis. Cases where pathogenic variants explaining the clinical phenotypes were detected are highlighted in the blue segment
The CHD7 pathogenic variants were categorized by genotype as follows: nonsense variants (n = 17) were the most frequent, followed by frameshift (n = 10), missense (n = 4), and splicing variants (n = 3). Two CNVs of CHD7 were identified through MLPA analysis, viz., an exon 3-38 deletion and a whole gene deletion. Four variants in CHD7, viz., c.1451del, c.2998dup, c.3424 G > T, and c.3522 + 1 G > T, have not been reported previously.
Exploring alternative genetic explanations for clinically suspected CHARGE syndrome in patients lacking CHD7 pathogenic variantsFor 23 patients who did not harbor pathogenic variants, further investigations were conducted, including karyotyping (n = 18), chromosome microarray (CMA, n = 9), ES (n = 7), and GS (n = 7).
Among 10 patients diagnosed with CHARGE syndrome according to either the Verloes or Hale criteria (Groups A–C), two different genetic diseases were identified in 2 patients (Supplementary Table 1).
Patient S40, a 1-year-old boy, was diagnosed with atypical CHARGE syndrome according to the Verloes criteria, but not the Hale criteria. He had intrauterine growth retardation (IUGR), macrocephaly, frontal bossing, short palpebral fissures, facial asymmetry, cleft palate, hypotonia, club feet, hypospadias, bilateral cryptorchidism, ventriculomegaly, duplex kidney, hearing loss, developmental delay, and congenital heart anomaly. Additional findings encompassed a choroid fissure cyst, hydrocephalus requiring endoscopic third ventriculostomy, hypothyroidism, and pigmented retinopathy. Single-gene testing for CHD7 showed normal findings, and his karyotype was 46,XY. At 2.7 years, CMA revealed a pathogenic 2.5-Mb deletion at 19q13.32-q13.33, comprising 99 genes, of which 63 are remarkable OMIM-listed genes.
Patient S18, a female born at full term with IUGR, was diagnosed with typical CHARGE syndrome according to both Verloes and Hale criteria. She had microcephaly, unilateral cataract, coloboma, anotia, ventriculomegaly, thinning of the corpus callosum, and congenital heart anomaly. GS revealed a previously unreported, de novo pathogenic variant in OTX2, c.728dup (p.Ala244Serfs*16), and her diagnosis was changed to microphthalmia, syndromic 5 (MCOPS5; OMIM#610125).
Identification of genetic background in patients sharing phenotype with CHARGE syndromeIn Group D, different genetic causes were identified in five patients (Supplementary Table 1).
Patient S51 had congenital heart anomaly, ear anomaly, facial palsy, right ptosis, frontal bossing, and hypotonia. She died due to progressive heart failure at the age of 4 months. Chromosome analysis revealed 46,XX,del(4)(p15.3) (Wolf–Hirschhorn syndrome; OMIM#194190).
Patient S52 had microcephaly, finger clinodactyly, hearing loss, agenesis of the corpus callosum, epilepsy, and developmental delay. ES revealed a 3-Mb deletion at 4p16.3 (Wolf–Hirschhorn syndrome; OMIM#194190).
Patient S53 showed frontal bossing, facial asymmetry, microphthalmia, low-set ears, webbed neck, hypertrophic cardiomyopathy, developmental delay, and short stature. ES revealed a de novo pathogenic variant, c.1924G>A (p.Asp642 Asn), in PLCB4 (auriculocondylar syndrome 2A; OMIM#614669).
Patient S56 had low-set ears, high-arched palate, short digital phalanges, congenital heart anomaly, multicystic dysplastic kidney, hearing loss, cryptorchidism, and hypoplastic inferior vermis of the cerebellum. GS revealed a de novo pathogenic variant, c.3316 G > A (p.Glu1106Lys), in TRRAP (developmental delay with or without dysmorphic facial features and autism) (DEDDFA; OMIM#618454).
Patient S59 presented hypertelorism, flat nasal root, micrognathia, high-arched palate, facial asymmetry, polydactyly, congenital heart anomaly, laryngomalacia, and feeding difficulty. She had only two minor features of CHARGE syndrome with a normal karyotype and no pathogenic variant in CHD7. CMA revealed a pathogenic 2.1-Mb deletion at 1q21.1-q21.2.
To summarize, this comprehensive genetic evaluation identified seven different genetic causes in 43 of 59 patients (72.9%), including CHD7 (36 patients, 61.0%) variants, Wolf–Hirschhorn syndrome (2 patients, 3.4%), 1q21.1 deletion (1 patient, 1.7%), 19q13 deletion (1 patient, 1.7%), OTX2 (1 patient, 1.7%), PLCB4 (1 patient, 1.7%), and TRRAP (1 patient, 1.7%) variant.
Comments (0)