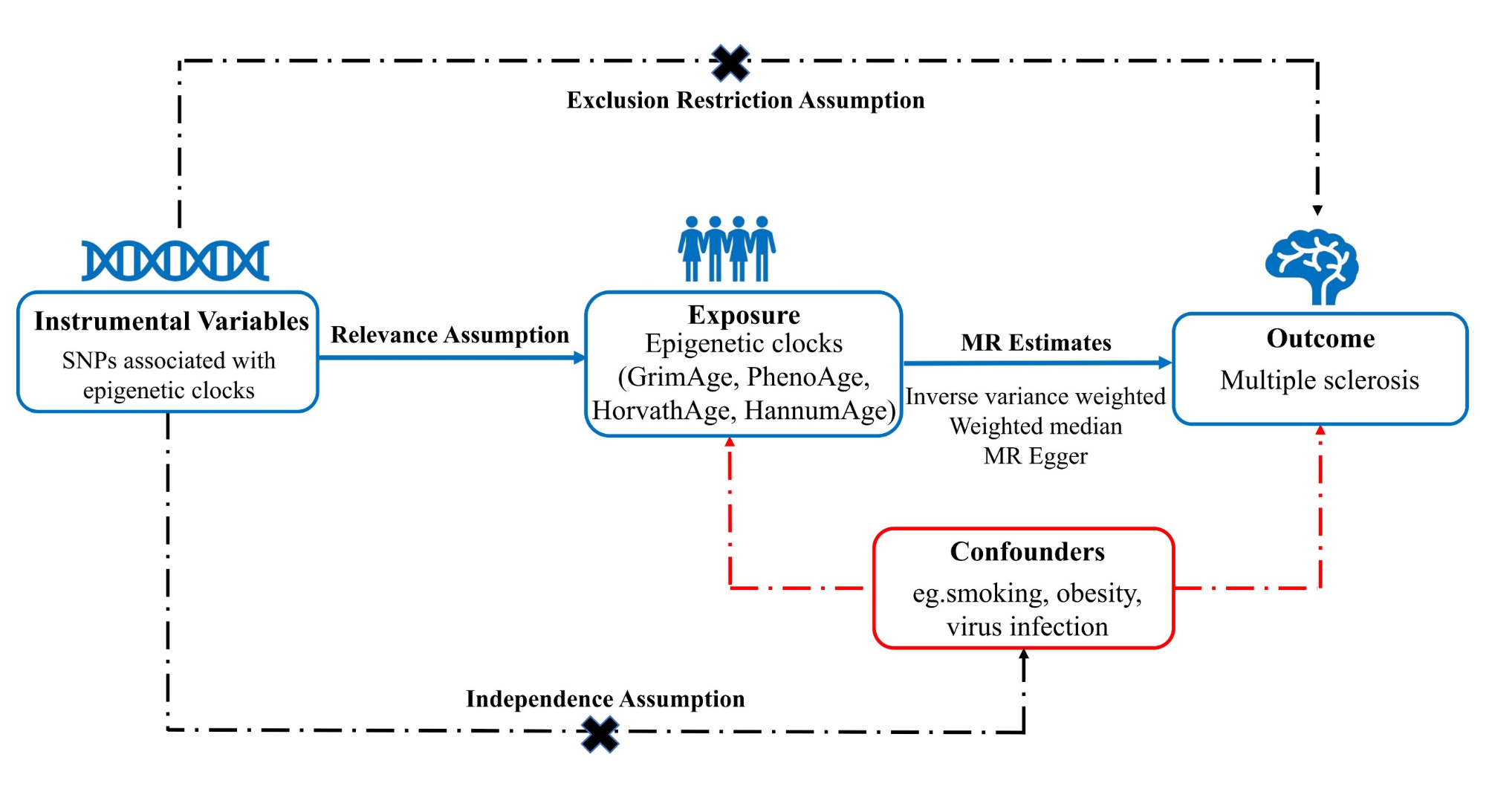
This is the first bidirectional causal relationship investigation between EAA and MS through a large-scale MR study, to the best of our knowledge. Our research reveals that an increased genetically predicted HannumAge is associated with a higher risk of MS. Conversely, we found no evidence supporting a causal effect of MS on any epigenetic aging-related traits. This suggests that EAA, specifically HannumAge, may be a risk factor for the development of MS.
Chronological age is calculated based on an individual’s birthdate, while biological age is assessed through biomarkers that gauge the aging process in various organs and tissues. Research has shown that biological age can diverge significantly among individuals of the same chronological age [35, 36]. DNA methylation at cytosine-phosphate-guanine (CpG) sites is a primary epigenetic marker that changes with age and can be measured in tissues and blood. The methylation percentage at each CpG site helps develop an “epigenetic clock,” which closely aligns with chronological age [6, 10]. Different epigenetic clock algorithms can be derived from methylation data across specific cell or tissue types, tailored to detect specific physiological changes or outcomes. The disparity between epigenetic and chronological ages can indicate whether individuals are biologically older or younger than their chronological age. Accelerated epigenetic aging is linked to a higher risk of age-related ailments, including neurodegenerative diseases like Parkinson’s and Alzheimer’s disease, cardiovascular diseases, and diabetes, as well as increased chances of early death [7, 37].
Previous research has established a link between DNA methylation and MS. Hypomethylation in genes, particularly those related to lymphocyte-mediated leukocyte and immunity pathways, has been shown to contribute to the immune-mediated pathology observed in MS [38]. Additionally, DNA methylation plays a role in modulating the immunogenicity of autoantigens in MS brain [39, 40]. Furthermore, in the peripheral blood mononuclear cells of MS patients, increased PAD2 expression coupled with promoter hypomethylation has been observed, highlighting the epigenetic alterations associated with the disease [41].
EAA may contribute to the development of MS through several interrelated mechanisms, as supported by existing research. 1) Immune dysregulation and “immunosenescence”: EAA is closely linked to age-related changes in immune function, collectively termed “immunosenescence;“ [42,43,44,45] these changes include diminished regulatory T cell efficacy and a shift towards pro-inflammatory immune phenotypes. Aberrant methylation patterns in genes regulating T cell differentiation and antigen presentation may amplify autoimmune responses, a hallmark of MS pathogenesis [42, 46]. 2) Neuroinflammation amplification: epigenetic modifications associated with EAA may upregulate pro-inflammatory cytokines like IL-6 and TNF-α; [47,48,49] these changes exacerbate microglial activation, impair the blood-brain barrier, and promote immune cell infiltration into the CNS, driving MS-related neuroinflammation [50, 51]. 3) Cellular senescence in neural and glial cells: accelerated biological aging may induce cellular senescence in key CNS cells, including oligodendrocytes and astrocytes; [42, 52, 53] senescent cells release inflammatory mediators, collectively known as the senescence-associated secretory phenotype, which hampers remyelination and exacerbates neural damage central to MS progression [54]. 4) Oxidative stress and mitochondrial dysfunction: EAA has been associated with oxidative stress and mitochondrial dysfunction, which are pivotal in MS pathology; [42, 55, 56] oxidative damage to myelin-producing cells and neurons drives disease progression, and epigenetic alterations may impair antioxidant defenses, exacerbating these processes [57]. 5) Environmental and Lifestyle Factors as Mediators: EAA serves as a cumulative marker for exposure to environmental and lifestyle factors, including stress, vitamin D deficiency, and poor diet; [58,59,60,61] these exposures are independently established as MS risk factors and may synergize with EAA to amplify autoimmune responses and promote disease development [62].
Early research indicates accelerated epigenetic age in individuals with MS. Theodoropoulou E and colleagues observed that the EAA of PhenoAge, which more accurately predicts aging outcomes compared to HorvathAge and HannumAge, was increased in whole blood samples from MS patients relative to healthy controls [63]. Similarly, Maltby et al. found GrimAge in the B cells of MS participants [12]. In contrast, Kular L and their team reported no significant increase in EAA across HorvathAge, GrimAge, PhenoAge, and HannumAge in the glial cells of MS patients compared to controls [13]. Furthermore, the same set of epigenetic clocks revealed significant disparities related to biological aging in lung immune cells influenced by MS and smoking [64]. These inconsistent findings might be due to the limited number of cases, the absence of longitudinal follow-up, and a lack of comprehensive analysis of MS outcomes. In this study, we investigated the causal link between EAA and MS using MR analyses. To reduce potential bias due to population stratification, we exclusively used GWAS data from individuals of European ancestry. Our data were carefully sourced from the Edinburgh DataShare and the International Multiple Sclerosis Genetics Consortium (IMSGC) Database, ensuring there was no overlap in samples. Quality control measures were implemented to verify the reliability and robustness of our results. This research enhances our understanding of MS risk factors, showing that increased odds of MS are associated with EAA as measured by the HannumAge clock. Epigenetic clocks hold promise as critical tools for clinicians and preventive medicine practitioners in assessing MS risk. Additionally, slowing down biological aging has become a significant area of interest in MS research.
In the context of our analysis, we utilized GrimAge, PhenoAge, HorvathAge, and HannumAge, which are widely recognized first-generation epigenetic clocks. These clocks were chosen for their robustness, extensive validation, and well-established associations with various age-related conditions, including neurodegenerative diseases like MS. Their widespread application highlights their reliability in capturing biological aging processes. Nonetheless, we recognize the potential of advanced epigenetic clocks, such as DamAge and AdaptAge, which represent significant advancements in the field. These newer clocks offer refined granularity and may capture more nuanced biological aging signatures, providing deeper insights into age-related pathologies like MS. Unfortunately, the dataset used in this study lacked the specific methylation markers required to calculate DamAge or AdaptAge scores, precluding their inclusion in the present analysis. Looking ahead, we are eager to incorporate DamAge and AdaptAge in future research. Their application, particularly in conjunction with longitudinal data and larger sample sizes, could greatly enhance our understanding of the intricate interplay between epigenetic age acceleration and MS pathogenesis.
GWAS data we used were drawn exclusively from populations of European ancestry, limiting the generalizability of our findings to other ethnic groups. This necessitates caution when extending these results to racially and ethnically diverse populations. Additionally, we relied on aggregate GWAS data, which prevented a stratified analysis by factors such as age and gender due to the absence of individual-level data. Despite efforts to mitigate confounding, we could not estimate the degree of overlap between exposure and outcome data in the two-sample MR analysis. To minimize bias from sample overlap, we employed robust instruments, ensuring an F statistic substantially greater than 25. Although we reduced confounding bias from SNPs, there remains the possibility that some SNPs could be linked to undetected factors that might affect the association between EAA and MS. The potential pleiotropic effects of these SNPs cannot be completely ruled out, warranting a cautious interpretation of our MR analysis results. Moreover, the epigenetic GWAS data we utilized are based on blood counts and clinical markers, which could lead to different outcomes as more diverse samples and GWAS data become available in the future.






Comments (0)