
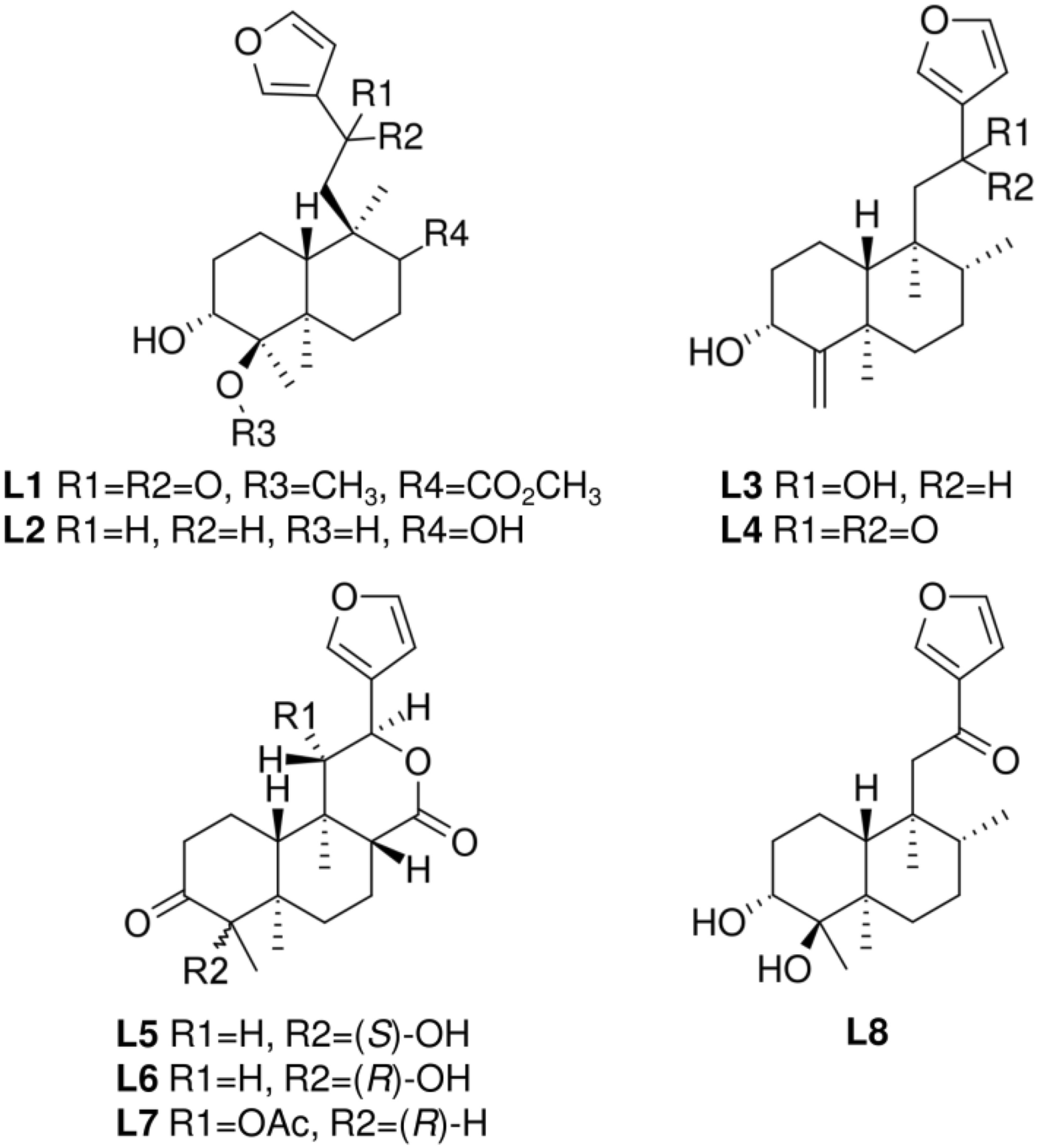
Remember me



The possible therapeutic action route of the L1-8 derivatives based on their chemical structure was identified through the application of the structure-based virtual screening (SBVS) protocol (Fig. 3). The ligands were observed to exhibit between 40 and 50% of their bioactivity as ligands for GPCRs, also known as membrane receptors, and proteins of the enzyme class, with the exception of the L5-6 derivatives, where this range can expand to 60–70% (Fig. 3A). The ligands can bind to drug transporters such as P-gp (in cell membrane) and plasma proteins (in systemic circulation), as shown by the blue colour bar in the graph in Fig. 3A. It is important to maintain objectivity and avoid subjective evaluations.
Fig. 3
Virtual structure-based screening results expressed as A percentage bioactivity of each L1-8 derivative by biological target class and B 3D similarity with known bioactive compounds from the ChEMBL database for identification of the specific biological target, where the colour spectrum ranges from red (few or no similar compounds) to blue (up to 240 similar bioactive molecules)
The ligands exhibited a high structural similarity with other mAChR M1 ligands in the ChEMBL database, particularly with oxygenated heterocyclic compounds. This class includes the L1-8 Laeviganoids (Fig. 3B), with the L1, L4 and L8 derivatives standing out due to their structural similarity with at least 114 compounds in the database that are mAChR M1 ligands. These derivatives are strong candidates against Tinnitus (Table 1). Furthermore, the compounds exhibited structural similarity with at least 32 bioactive compounds found in the ChEMBL database that serve as substrates for the CYP450 isoform type 17A1 (CYP17A1) during phase I metabolism (refer to Table 1). The heatmap in Fig. 3B shows that the majority of the compounds (represented by white to blue spectra) undergo biotransformation in the human liver microsome system, except for derivatives L2 and L7 (represented by red spectra), indicating that these substances are not biotransformed in pre-systemic metabolism.
Table 1 Number of similar compounds (known 3D structures) to the L1-8 derivatives for different specific targets with a considerable number of similar compoundsTopological analysis and druglikenessIn the topological analysis of the MLP shown in Fig. 4, it is evident that the polar oxygenated H-bond donor and acceptor groups (OH and O groups) have an impact on the molecule’s surface accessibility to both aqueous and organic environments of the L1-8 derivatives. In this analysis, it was observed that the L4 ligand had the highest AlogP value of 4.62 (Table 2). This was due to its monohydroxylated fused bicyclic structure (blue to green colour spectra) and polar H-bond acceptor contribution in the portion of the carbonyl linked to the furan (yellow to red colour spectra), compared to the dihydroxylated L2 derivative.
Fig. 4
Ligands L1-8 with their molecular lipophilicity potentials (MLP) plotted and analysed in terms of the polarity of the molecular fragments, where the colour spectrum ranges from red (polar groups) to blue (hydrophobic or apolar groups), resulting in the calculated values of AlogP
Table 2 Physicochemical properties of lipophilicity (AlogP), molecular weight (MW in g/mol) and polarity (TPSA in Å2) calculated and applied to the GSK and Pfizer biopharmaceutical classification systemsOn the other hand, the derivatives L5-6 exhibited an AlogP value of 2.91 (Table 2), indicating moderate lipophilicity. The difference in the position of the substituted (R) and (S)-OH groups (Fig. 1) did not affect the distribution of the hydrophilic surface. The hydrophilic surface showed a strong contribution from the carbonyl groups of the fused tricyclic portion of these derivatives (red colour spectra), as shown in Fig. 4.
When considering the polarity values (TPSA), it is evident that the lipophilicity of the ligands has a significant impact on the distribution of the compounds in the CNS [45]. The L4 derivative, with a TPSA of approximately 50, is the least polar. The value of 44 Å2 reflects the contribution of the polar surfaces of the carbonyl portion conjugated to the furan ring. The H-bond acceptor groups of the C = O and R-O-R type are less polar than OH-type donor groups [46]. This results in a low susceptibility to being a P-gp substrate, as indicated by the red colour in Fig. 5. Therefore, this compound is more effective at the CNS than the other analogues [47]. Compound L4 scored positively ( +) for the Pfizer rule, indicating that it resides in a physicochemical space formed by compounds of high lipophilicity (AlogP > 3) and low polar surface area (TPSA < 75 Å2), suggesting distribution to the CNS [25]. However, the GSK rule raises a structural concern regarding its lipophilicity, which may limit its pharmacokinetics (Table 2). Furthermore, all derivatives possess an optimal polarity for effective gastrointestinal absorption due to the presence of oxygenated groups such as R-OH and R2O [48]. The estimated bioavailability (%F) is unlikely to be less than 30% as shown in Table 3.
Fig. 5
Alignment graph between lipophilicity (AlogP) and polar surface (TPSA) for the estimation of the BBB activity, where the red region is the physicochemical space of access to the CNS, formed by compounds with high AlogP and low TPSA, and the region outlined up to TPSA = 75 Å2 is the region CNS safety. The blue indicators are compounds whose presence of OH groups results in a polarity that makes the molecules susceptible to efflux by P-gp, while the red indicators highlight passively permeable molecules
Table 3 Pharmacokinetic descriptors predicted by the consensus test of ADME properties between the SwissADME, ADMETlab 2.0 and AI Drug Lab databasesAlthough this step constitutes an empirical prediction of druglikeness, pharmacokinetic descriptors were provided to corroborate the results, thereby ensuring greater alignment between the predictive techniques. The results of the pharmacokinetic prediction demonstrate a strong correlation with the physicochemical attributes evaluated in this section, which can be seen in Table 3.
Predicting ADME propertiesThe predictive analysis of ADME and the topological analyses of druglikeness based on the chemical structure of the L1-8 derivatives are in agreement. It was observed that compounds located in a physicochemical space defined by AlogP > 3 and TPSA ≤ 75 Å2 may exhibit activity in the CNS. Here, compound L4 is notable for its low polarity, which results in greater cellular permeability compared to efflux.
In this way, the parallel artificial membrane permeability assay (PAMPA) descriptors were employed to estimate the cell permeability potential of the L1-8 derivatives [23, 49]. The results demonstrated that the pharmacokinetic properties align with the physicochemical and structural trends observed in the compounds. This supports the idea that the compound is unlikely to be a P-gp substrate, as well as the Papp,A→B descriptor of 1.9 × 10–5 cm/s, this suggests that the compound can penetrate more selective cell membranes, such as the MDCK cell model, commonly employed to estimate the permeability of active drug candidates in the central nervous system (CNS). The L2, L6, and L8 compounds exhibited Papp,A→B MDCK values exceeding 2.0 × 10⁻5 cm/s, yet they may function as P-gp substrates. This suggests they may undergo passive efflux to the extracellular environment, potentially reducing the viability of small molecules acting in the CNS. Although derivatives L1 and L7 exhibited a high predicted apparent permeability coefficient (Papp,A→B MDCK in the order of 5.6 and 2.1 × 10⁻5 cm/s, respectively), the compounds may be highly polar as a consequence of the substituted carboxyl-based groups (R-COO-R) in the Laeviganoid substructure (Fig. 5).
Furthermore, it was observed that the compounds exhibited a lipophilicity range indicative of high hydrophobicity, which may facilitate the formation of chemical entities widely distributed in the organic phase of physiological environments. It was observed that compound L4, likely to be active in the CNS without being a P-gp substrate, exhibited a predicted relative Vdss value of 4.91 L/kg. This indicates that the high lipophilicity of the compound facilitates a broader distribution of the bioavailable molecular fraction in biological tissues than in blood plasma, thereby corroborating the likelihood of the substance’s access to the CNS, where the blood–brain barrier (BBB) is a highly selective biological membrane. Moreover, all the compounds exhibited Vdss values between 3.0 and 6.7 L/kg, indicating their high lipophilicity.
These findings are by the PPB prediction, whereby the estimated values below 55% indicate that a substantial proportion of the molecular fraction is not bound to the serum proteins of the blood plasma, thereby allowing for a more widespread distribution and the potential for a more significant biological effect. Furthermore, the predicted clearance rate (CLint,u) of 7.70 mL/min/kg is a significant factor influencing the oral bioavailability of the L4 derivative. This suggests that the compound may exhibit metabolic stability at the level of hepatic microsomes, resulting in a high concentration of the non-biotransformed molecular fraction in the systemic circulation (Table 3). The compound’s metabolic stability is directly related to the formation of low-reactive metabolites in the human liver microsome system. This can be observed in the section on the site of metabolism (Fig. 6).
Fig. 6
Prediction of the site of metabolism of the derivatives A L1, B L2, C L3 and D L4, considering the probability that they are substrates of the CYP450 isoforms in phase I metabolism
Site of metabolism predictionThe test for structure-dependent metabolism site prediction allowed for the estimation of potential secondary metabolites resulting from the oral administration of L1-8 derivatives. This test aligns the descriptors of structural specificity and sensitivity of the fragments with CYP450 substrate isoforms [27].
The observation made here is that the furan ring has unsaturated sites that are susceptible to aromatic hydroxylation (Fig. 6). This process can lead to the formation of reactive epoxide intermediates that are capable of intercalating into protein and DNA structures, as shown in Fig. 6. Notably, the ligands L3 (Fig. 6C) and L4 (Fig. 6D) feature an isolated alkene within the bicyclic ring of the Laeviganoids substructure. The presence of an aliphatic hydroxyl group at this position results in the formation of hydroxylated metabolites that exhibit reduced reactivity compared to the epoxides derived from the hydroxylation of the furan ring. This finding supports the hypothesis that the compounds in question have a low probability of causing liver damage or the formation of reactive metabolites that could interact with proteins and DNA (Table 3).
These fragments were identified through a test for detecting substrates of CYP450 isoforms in pre-systemic metabolism (Phase I). This test is based on structure–activity relationship (SAR) descriptors, which relate the structural specificity and sensitivity of the molecular fragment to its capacity to act as a CYP450 substrate. These are derivatives that can be derived from specific isoforms, such as CYP17A1, identified in the virtual screening of the target prediction stage. Consequently, they are processes that can reduce the systemic bioavailability of these substances. However, they do not negatively affect the order of hepatic clearance, leading to the formation of chemical species of low toxicity by metabolic activation (Table 3).
Molecular docking against mAChR M1At the end of the cycle of 50 independent molecular docking simulations, each consisting of 20 poses for each of the ligands, it was observed that all the ligands achieved a statistical adjustment of RMSD of less than 2.0 Å. This indicates optimal specificity for the amino acid residues of their binding sites on the mAChR M1 (Fig. 7A). The redocking carried out with the inhibitor TTP achieved an RMSD of around 0.99 Å, which was used as the statistical standard for the other simulations (orange bar). The L4 ligand formed a ligand–receptor complex with mAChR M1 with an affinity energy of approximately −8.7 kcal/mol, which meets the ideality standard (affinity energy < −6.0 kcal/mol) and provides more favourable energy conditions than the TTP inhibitor (−7.157 kcal/mol) (Fig. 7B).
Fig. 7
Data from the molecular docking simulations expressed as A RMSD in Å and B affinity energy in kcal/mol, for the L1-8 derivatives and the TTP inhibitor against the mAChR M1
This affinity energy analysis is of great biological importance in understanding the biochemical interactions of small ligands with receptors expressed in the CNS. According to Shityakov et al., (2014) [50], the formation of ligand–protein complexes with an affinity energy lower than −6.0 kcal/mol is associated with CNS-active compounds that readily penetrate the BBB. This analysis suggests that compound L4 is a lead compound due to its CNS activity characteristics and affinity energy against the mAChR M1 receptor (Fig. 7B).
When analysing the ligand–receptor interactions, it was possible to observe that the L4 ligand complexed to the mAChR M1 interacting with amino acid residues in common with the TTP inhibitor, revealing that the compound binds to the same interaction site as the co-crystallised inhibitor (Fig. 8A). Here, it was possible to observe that the L4 ligand showed hydrophobic interactions in common with the TTP inhibitor, including interactions with the aromatic side chains of the Tyr106, Trp378, Tyr381 and Tyr404 residues (Fig. 8B), where the calculated distances between the ligand and these amino acid residues are around 3.1–3.4 Å (Table 4), characterising interactions of moderate strength (blue to white spectra in Fig. 8C) [51]. It is interesting to note that the inhibitor interacts with the aromatic centre of the Tyr106 residue by hydrophobic interaction with a contribution from the π electrons of its thiophenyl portion (Fig. 8D), while the L4 derivative acts as an H-bond acceptor for the hydroxyl of the residue through its furanyl portion (Fig. 8E).
Fig. 8
A Three-dimensional representation of mAChR M1 with the ligands TTP and L4 in their binding sites, B interactions between the ligands and the residues of the receptor’s active site, C heatmap relating the distances between the ligands and the amino acid residues to the interaction forces, where the colour spectrum ranges from blue (strong interactions) to red (weak interactions) and two-dimensional representations of the structural contributions of the ligands D TTP and E L4 in the interactions with the residues of the receptor’s active site
Table 4 General data on the interactions between the L1-8 derivatives and the mAChR M1, compared to the TTP and BZP controls, expressed as types of interaction, interaction residues and ligand–receptor distance involved in each type of interaction characterised in the molecular docking simulationsOn the other hand, the other ligands (L1-3, and L5-8) bound to mAChR M1 at a site believed to be the allosteric modulation site of the benzodiazepine BZP. They interact mainly with the aromatic residues Tyr82, Thr189, Tyr381, Trp400 and Tyr404 (Fig. 9A). Here, interactions of similar strength between ligands L1-3, L5-6, and L8 with the aromatic side chain of the Tyr404 residue are highlighted. The distances calculated between 3.0 and 4.0 Å reveal interactions of moderate to weak strength (clear blue to white spectra in Fig. 9B).
Fig. 9
A Three-dimensional model of the coupling of the L1-3 and L5-8 derivatives to the BZP allosteric modulation site in relation to the TTP inhibitor B heatmap relating the distances between the ligands and the amino acid residues to the interaction forces, where the colour spectrum varies from blue (strong interactions) to red (weak interactions)
Molecular dynamics simulationsRMSD analysisFollowing an analysis of the Molecular Dynamics (MD) simulations, Fig. 10 presents the results of the root-mean-square deviation (RMSD) variations of the simulated systems, with duplicates classified as Run 1 (black) and Run 2 (red).
Fig. 10
RMSDMD variations of the simulated systems A M1 receptor B System formed in the presence of the inhibitor TTP C System composed of the ligand L4
Figure 10A shows the variations of the biological receptor without ligands in its cavities. The RMSD data show that the two simulations are similar. In the first simulation (black), between 0 and 17 ns, the RMSD variations were around 2.5 Å. From 18 ns onwards, the RMSD values of both simulations remained between 2.8 Å and 3.1 Å until the end of the simulation. This behaviour indicates that the receiver showed minor structural deformation. In contrast, in the second run associated with the M1 receptor, there was a more pronounced structural variation with values of 4.6 Å at 51 ns and a further deformation of 4.3 Å observed at approximately 73 ns. After this point, the system showed minor structural variations and remained cohesive until the end of the simulation, with values of around 3.0 Å.
The variations of the system with the TTP inhibitor associated with the M1 receptor are shown in Fig. 10B. Both runs showed values of around 2.5 Å up to 20 ns. After this period, up to 38 ns, the first run increased structural deformation, reaching 4.2 Å, while the second run maintained values close to 2.6 Å. After 40 ns, the RMSD values of the two runs remained relatively constant and similar, ending the simulation with values of 2.6 Å and 3.0 Å for Run 1 and Run 2, respectively.
In Fig. 10C, the results of the simulations with the L4 ligand inserted show that both runs showed similar behaviour in terms of structural deformation of the system. In the first run, the RMSD variations remained around 2.8 Å up to 40 ns and remained constant until the end of the simulation, reaching around 4.0 Å at 200 ns. The second run showed pronounced oscillations, reaching values of up to 4.2 Å at certain times and remaining consistent until 88 ns, with deformations of around 3.4 Å. Between 90 and 100 ns, the second run showed pronounced deformation, reaching 4.3 Å. After 100 ns, the complex with L4 showed less conformational variation, remaining relatively stable until the end of the simulation, with values around 3.8 Å.
These results show the consistency and relative stability of the MD simulations for the different systems analysed, indicating the conformational variations with and without the presence of the ligands.
According to the data from the MD simulations, the system with the M1 receptor alone showed minor conformational variations, reaching RMSD values of up to 4.6 Å, indicating low structural variation. When the inhibitor TTP was added to the M1 receptor, the results showed a progressive increase and more coherent values over the 200 ns of the simulation. After approximately 140 ns, the system showed stability with less structural deformation, maintaining values close to 4.2 Å.
In the simulation with the L4 ligand, the structural variations were similar to those observed with the TTP inhibitor, reaching values of up to 4.3 Å. These data suggest that the L4 ligand can induce a structural deformation in the M1 receptor similar in magnitude to that caused by the TTP inhibitor. Thus, the presence of the L4 ligand appears to play a comparable role in the structural stability of the receptor, as indicated by the analysis of the RMSD deviations.
Hydrogen bond analysisThe results of the MD prediction simulations were used to evaluate the percentage of hydrogen bond formation between the initial (0 ns) and final (200 ns) states of the simulation. Interactions with amino acid residues in the inhibition site (Asp105, Tyr106, Trp157, Ala196, Val113, Phe197, Asn382, and Trp378) were considered as criteria for analysing favourable interactions to determine an interaction profile and estimate the viability of the compounds analysed. Only contributions greater than or equal to 5% of the total simulation were included for the analysis of hydrogen bond formation.
Figure 11A shows the percentage of hydrogen bonds formed between the TTP ligand and the M1 receptor. The results show that throughout the simulation, TTP interacted with 13 amino acid residues in the receptor, the most important being Tyr404 (55.34%), Asn382 (42.61%), Cys407 (29.72%), Tyr106 (17.23%), and Ala196 (13.24%). Other residues were Phe197 (8.49%), Tyr408 (8.54%), Tyr381 (12.79%), Asp105 (5.04%), Gln110 (5.45%), Trp378 (8.19%), Trp157 (6.54%) and Thr192 (6.99%).
Fig. 11
Percentage of hydrogen bond formation A system composed of the TTP/M1 complex B system formed in the presence of the L4/M1 complex
In Fig. 11B, the data for the system with the ligand L4 associated with the M1 receptor show that L4 formed hydrogen bonds with ten amino acid residues, most frequently with residues Tyr106 (71.03%), Trp378 (35.57%), Ala196 (19.03%) and Tyr381 (16.99%). Other residues involved were Ser109 (15.93%), Asn382 (13.64%), Phe197 (11.29%), Thr192 (6.95%), Leu183 (6.75%) and Trp157 (5.85%).
These results provide an overview of the hydrogen interactions between the ligands and the M1 receptor and highlight the amino acid residues that play a crucial role in stabilising the TTP/M1 and L4/M1 complexes throughout the simulation.
The molecular dynamics simulations indicated that both ligands formed hydrogen bonds with residues in the inhibition site of the target receptor. The TTP ligand established hydrogen bonds with seven amino acid residues in this site (Asp105, Tyr106, Trp157, Ala196, Phe197, Asn382 and Trp378), with binding percentages ranging from 5.04 to 42.61%. These findings indicate that TTP exhibits high specificity for the receptor cavity and maintains its presence in the interaction site as predicted by molecular docking simulations, thereby substantiating its potential as an inhibitor.
The L4 ligand demonstrated the formation of hydrogen bonds with five amino acid residues within the inhibition site (Trp157, Ala196, Asn382, Tyr106 and Phe197), with frequencies ranging from 5.85 to 71.03%. These values indicate that L4 also exhibits high specificity for the predicted cavity, thereby substantiating its viability as an M1 receptor inhibitor, in a manner analogous to TTP.
These findings underscore the potential of both ligands as M1 receptor inhibitors, exhibiting structural specificities that align with the predictions derived from molecular docking.
MM/GBSA analysisThe results of the molecular dynamics simulations indicated which complex systems (ligand and receptor) exhibited the most favourable free energy indices, as determined by ΔGbind (kcal/mol) through the MM/GBSA calculations. These findings are detailed in Table 5. The free energy values for the TTP/M1 and L4/M1 systems were -10.72 ± 2.78 kcal/mol and −26.93 ± 2.96 kcal/mol, respectively. In the TTP/M1 system, the terms with the greatest contributions to the free energy of binding were EvdW and Eele, with values of −53.90 kcal/mol and 41.79 kcal/mol, respectively. In the L4/M1 system, the most significant contributions came from EvdW and GGB, with values of −38.95 kcal/mol and 21.26 kcal/mol, respectively.
Table 5 Free energy values associated with the TTP/M1 and L4/M1 complexesThese values highlight the most relevant interactions and suggest greater stability for the L4/M1 system in comparison to TTP/M1, as evidenced by the differences in free energy of binding between the complexes.
The TTP/M1 complex exhibited three favourable contributions to the free energy of binding, derived from the EvdW, GGB and GSA terms. Conversely, the Eele and -TΔS terms were identified as unfavourable contributions. For the L4/M1 complex, the EvdW, Eele and GSA terms enhanced the affinity energy, whereas the GGB and -TΔS terms exerted unfavourable effects.
The results of the molecular dynamics simulations indicate that both systems are viable for interaction, with binding free energy values of −10.72 ± 2.78 kcal/mol for TTP/M1 and −26.93 ± 2.96 kcal/mol for L4/M1. A more detailed analysis revealed that the L4/M1 complex exhibited greater viability of interaction due to its lower binding free energy value, indicating enhanced stability for this system.
Comments (0)