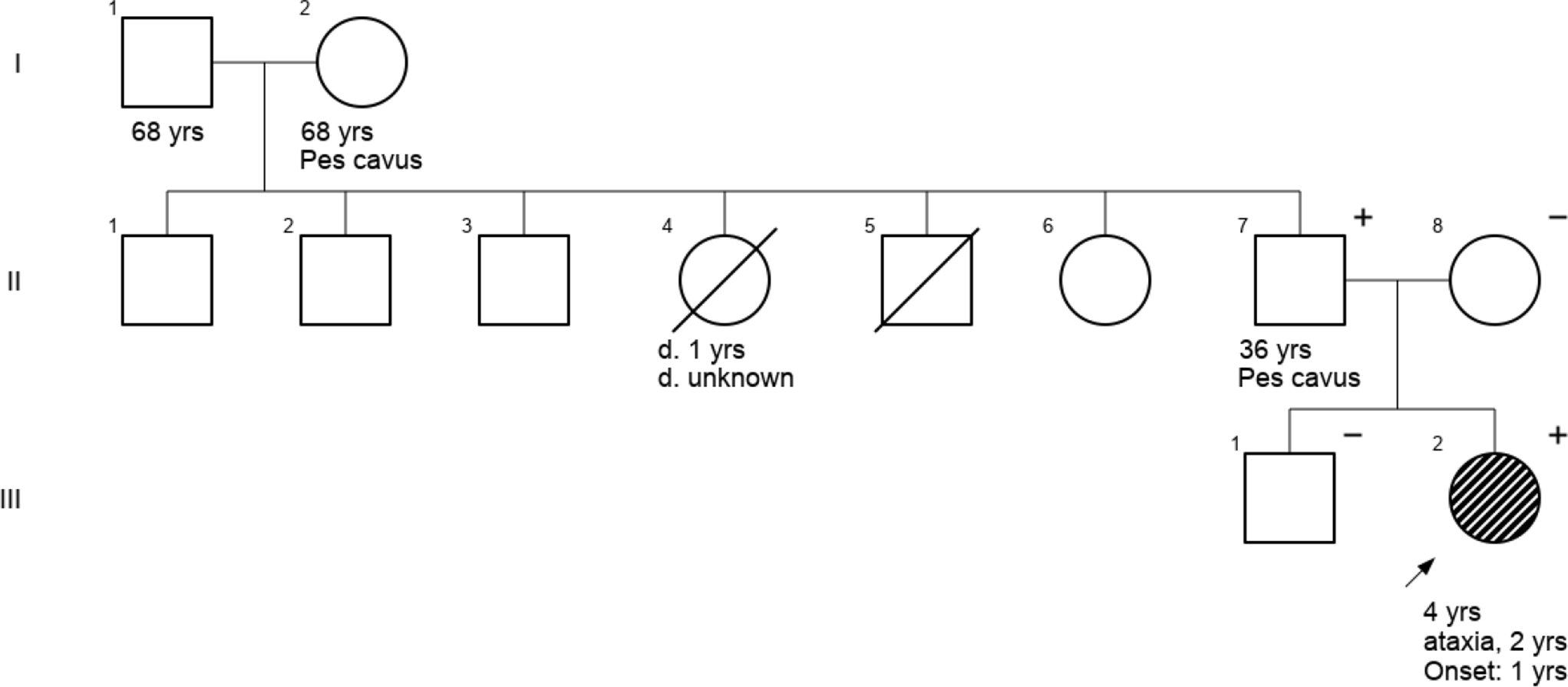
We here describe a SCA27B patient with a proven pathogenic FGF14 repeat expansion whose symptoms were initially considered as post-surgical sequelae that deteriorated due to a collapse of compensatory mechanisms with advanced age. Her progressive cerebellar symptoms, the presence of DBN, further family details, and cerebellar atrophy extending beyond the lesional area prompted additional genetic testing, which eventually identified a FGF14 repeat expansion as the major driver of her progressive cerebellar symptoms over the past four years.
The key triad of SCA27B is an age at onset after 45 years, episodic symptoms, and DBN [3]. Non-cerebellar symptoms can include vestibular areflexia and peripheral axonal sensory neuropathy, dysautonomia, spasticity, and parkinsonian features [4]. Full penetrance of the mutation is observed in individuals with GAA> 300 expansions, while incomplete penetrance occurs with GAA250 − 300 with indications that this threshold might even be lower [1]. Heterozygous FGF14 repeat expansions are reported to be a frequent cause of LOCA, with frequencies up to 30% in European cohorts, but a lower frequency of approximately 1% in East-Asian cohorts [1, 3, 5, 6].
Oculomotor disturbances are a key component of the SCA27B phenotype, and have been reported in approximately 95% in a French cohort of SCA27B cases, with DBN being the most commonly reported oculomotor sign in up to 66% of their cohort [2, 3]. Although DBN is not exclusive to SCA27B, its presence combined with cerebellar ataxia should trigger testing for this disorder.
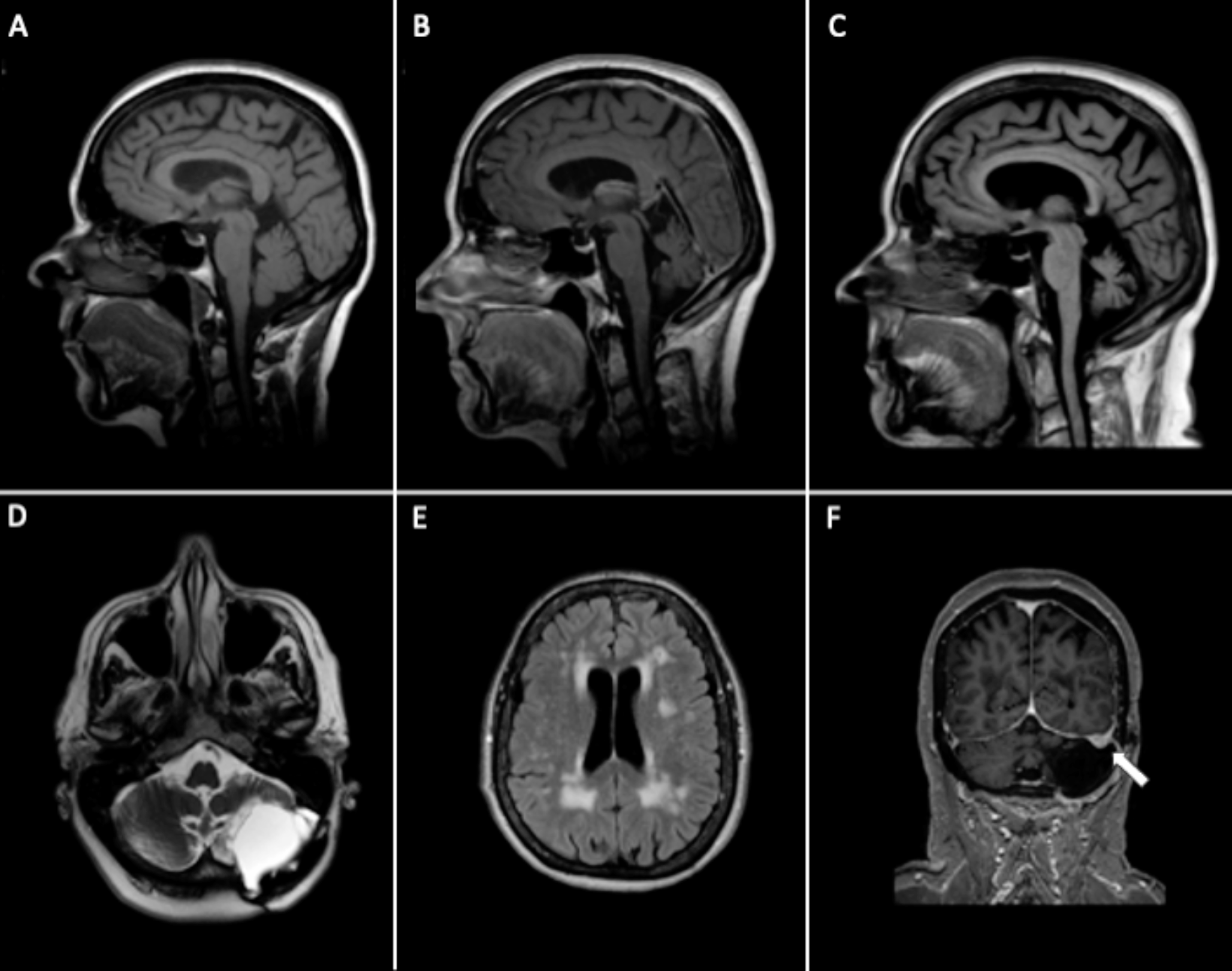
Cerebellar atrophy on brain imaging, predominantly affecting the vermian region, is frequently observed in SCA27B patients, although it is not invariably present [1]. Mild diffuse cerebellar atrophy was also observed in our case, along with extensive periventricular and brainstem WMA. Periventricular WMA were reported in 80% of patient with SCA27B, but the WMA in our case are best compatible with small vessel disease. Interestingly, involvement of superior cerebellar peduncles and the decussation within the midbrain is observed in up to 62% of SCA27B cases, which helps differentiating SCA27B from other LOCA subtypes [7]. These midbrain signal changes were absent in our patient.
The presence of post-surgical cerebellar lesions and residual ataxia led to an initial hypothesis of progression of these residual signs upon ageing. Re-emergence of previous deficits (or recrudescence) triggered by toxic or metabolic factors is a well-known phenomenon in stroke patients, although it is generally transient [8]. The subacute deterioration of cerebellar symptoms after a delay of twelve years, which appeared to be non-transient and progressive, plus the DBN and cerebellar atrophy without signs of new acquired causes, prompted us to consider genetic causes. At the time of initial genetic tests, testing for SCA27B was not yet available.
Our case underlines the importance of considering SCA27B in slowly progressive cerebellar ataxia with oculomotor abnormalities, regardless of the presence of a static cerebellar lesion. We recommend prioritizing genetic screening for SCA27B as the first diagnostic step in these patients; if negative, genetic testing should be expanded to what is routinely done for late-onset ataxias.




Comments (0)